
- Склад
- Лікарська форма
- Фармакотерапевтична група
- Фармакологічні властивості
- Клінічні характеристики
- Показання
- Протипоказання
- Взаємодія з іншими засобами
- Особливості застосування
- У період вагітності
- При керуванні автомобілем
- Спосіб застосування та дози
- Діти
- Передозування
- Побічні реакції
- Термін придатності
- Умови зберігання
- Упаковка
- Категорія відпуску
- Виробник
- Місцезнаходження виробника
Увага! Срок дії реєстраційного посвідчення UA/16651/01/01, UA/16651/01/02 закінчився 26.04.2023
Варгатеф інструкція із застосування
Офіційна інструкція лікарського засобу Варгатеф капсули 100 мг, 150 мг. Опис та застосування Vargatef, аналоги та відгуки. Інструкція Варгатеф капсули затверджена виробником.
Склад
діюча речовина: нінтеданіб; 1 капсула м’яка містить 100 мг або 150 мг нінтеданібу (у вигляді езилату);
допоміжні речовини: тригліцериди середньоланцюгові, твердий жир, лецитин (соєвий) (Е 322);
оболонка капсули: желатин, гліцерин 85%, титану діоксид (Е 171), заліза оксид червоний (Е 172), заліза оксид жовтий (Е 172); чорнило чорного кольору для маркування капсул: шелак, етанол, пропіленгліколь (Е 1520), заліза оксид чорний (Е 172).
Лікарська форма
Капсули м’які.
Основні фізико-хімічні властивості:
ВАРГАТЕФ. капсули м’які по 100 мг
Продовгуваті непрозорі м’які желатинові капсули персикового кольору, з одного боку чорним чорнилом нанесений логотип компанії «Boehringer Ingelheim» і маркування «100». Капсули містять в’язку суспензію яскраво-жовтого кольору.
ВАРГАТЕФ. капсули м’які по 150 мг
Продовгуваті непрозорі м’які желатинові капсули коричневого кольору, з одного боку чорним чорнилом нанесений логотип компанії «Boehringer Ingelheim» і маркування «150». Капсули містять в’язку суспензію яскраво-жовтого кольору.
Фармакотерапевтична група
Антинеопластичні засоби. Інгібітори протеїнкінази.
Код ATX L01X Е31.
Фармакологічні властивості
Фармакодинаміка
Механізм дії Нінтеданіб є інгібітором ангіокінази потрійної дії, який блокує рецептори фактору роста ендотелію судин (VEGFR 1-3), рецепторів тромбоцитарного фактору роста (PDGFR а та ß), рецептори фактору роста фібробластів (FGFR 1-3), через які здійснюється активність кінази. Нінтеданіб конкурентно взаємодіє з аденозинтрифосфат (АТФ)-зв’язуючою ділянкою цих рецепторів і блокує внутрішньоклітинну передачу сигналів, яка важлива для проліферації та виживання ендотеліальних. а також периваскулярних клітин (перицитів і гладком’язових клітин судин). Крім того, нінтеданіб інгібує кінази Flt-3 (Fms-подібна білкова тирозинкіназа), Lek (лімфоцит-специфічна білкова тирозинкіназа), Lyn (білкова тирозинкіназа Іуп) та Src (протоонкогенна білкова тирозинкіназа Src).
Фармакодинаміка
Пухлинний ангіогенез — це особливий процес, що сприяє росту пухлини, прогресуванню захворювання та утворенню метастазів. Цей процес переважно запускається проангіогенними факторами, які секретуються пухлинними клітинами (а саме, VEGF і bFGF), щоб залучити ендотеліальні і периваскулярні клітини хазяїна і полегшити доставку кисню і поживних речовин через судинну систему хазяїна. У доклінічних моделях захворювання нінтеданіб в якості монотерагіії ефективно протидіяв утворенню і розвитку судинної системи пухлини, що призводило до уповільнення і зупинки росту пухлини. Зокрема, лікування ксенотрансплантатів пухлини нінтеданібом призводило до швидкого зменшення щільності мікросудин пухлини, покриття перицитами судин та перфузії пухлини. Вимірювання методом динамічної контрастної магнітно-резонансної томографії показали антиангіогенний ефект нінтеданібу у людей. Він не повністю залежав від дози, але більшість реакцій спостерігалась при дозах >200 мг. Логістична регресія виявила статистично значущий зв’язок ангіогенного ефекту з експозицією нінтеданібу. Ефект динамічної контрастної магнітно-резонансної томографії спостерігався через 24 — 48 годин після першого прийому лікарського засобу та був збережений або навіть збільшений після тривалого лікування протягом кількох тижнів. Не встановлено взаємозв’язку відповіді, виявленої методом динамічної контрастної магнітно-резонансної томографії, та наступного клінічно значущого зниження розміру ураження мішені, але відповідь, виявлена методом динамічної контрастної магнітно-резонансної томографії, була пов’язана із стабілізацією захворювання.
Абсорбція Максимальна концентрація нінтеданібу в плазмі крові досягається приблизно через 2-4 години після перорального прийому препарату у формі м’яких желатинових капсул під час їди (діапазон 0,5-8 годин). Абсолютна біодоступність дози 100 мг складає у здорових добровольців 4,69% (90% ДІ: 3,615-6,078). Абсорбція та біодоступність зменшуються внаслідок дії транспортера та суттєвого пресистемного метаболізму. Встановлено, що експозиція нінтеданібу збільшується пропорційно дозі (у діапазонах доз 50^450 мг один раз на добу та 150-300 мг двічі на добу).
Стійкі концентрації в плазмі крові досягаються, як максимум, впродовж одного тижня після початку прийому. Експозиція нінтеданібу збільшується після їди приблизно на 20% у порівнянні з прийомом препарату натще (ДІ: 95,3-152,5%), а всмоктування сповільнюється (медіана часу досягнення максимальної концентрації в плазмі крові натще (tmax) — 2.00 години: після їди -3.98 години). Розподіл Розподіл нінтеданібу здійснюється шляхом двофазної кінетики. Після внутрішньовенної інфузії спостерігається великий об’єм розподілу (Vss): 1050 л, геометричний коефіцієнт варіації (gCV) 45.0%). Зв’язування нінтеданібу з білками плазми людини in vitro вважається значним, пов’язана фракція складає 97.8%. Основним білком, що бере участь в зв’язуванні, вважається альбумін сироватки крові. Нінтеданіб переважно розподіляється в плазмі, співвідношення кров/плазма складає 0.869, Біотрансформація Основною реакцією, що бере участь в метаболізмі нінтеданібу, є гідролітичне розщеплювання за допомогою естераз. що призводить до утворення вільного кислого метаболіту нінтеданібу (BIBF 1202). Надалі BIBF 1202 глюкуронізується ферментами уридін-5′-дифосфат-глюкуронозилтрасферази (UGT). а саме UGT 1А1, UGT 1А7, UGT 1А8 та UGT 1А10, з утворенням глюкуроніду BIBF 1202.
Біотрансформація нінтеданібу за участю ізоферментів CYP відбувається лише незначною мірою; основну роль у цьому процесі відіграє ізофермент CYP ЗА4. У дослідженні ADME у людини основний метаболіт, що утворюється за участю ізоферментів CYP. виявити в плазмі не вдалося. За даними дослідження in vitro CYP-залежний метаболізм складає приблизно 5%, тоді як розщеплювання, здійснюване естеразами, складає 25%. В доклінічних дослідженнях in vivo BIBF 1202 не показав ефективність, незважаючи на його активність на цільових рецепторах субстанції. Загальний кліренс плазми після внутрішньовенної інфузії є високим (СІ: 1390 мл/хв, gCV — 28.8%). Виведення із сечею незміненої активної речовини впродовж 48 годин після прийому нінтеданібу внутрішньо складає приблизно 0.05% від величини дози (gCV — 31.5%), а після внутрішньовенного введення — приблизно 1.4% (gCV — 24,2%); нирковий кліренс складає 20 мл/хв (gCV — 32,6%). Після прийому внутрішньо [І4С]-нінтеданібу радіоактивний матеріал виводився переважно з жовчю і виявлявся в калі (93.4% дози. gCV — 2.61%). Частка ниркової екскреції в загальному кліренсі є низькою (0.649% дози (gCV — 26,3%)). Виведення вважається повним (більше 90%) через 4 дні після прийому. Період напіввиведення нінтеданібу в термінальній стадії складає від 10 до 15 годин (gCV приблизно 50%).
Лінійність/нелінійність Можна припустити, що фармакокінетика (ФК) нінтеданібу лінійна відносно часу (тобто дані щодо застосування разової дози можуть бути екстрапольовані на дані щодо багаторазового використання). Значення Стах в результаті накопичення препарат) після багаторазового застосування перевищує показник Сщах разової дози в 1,04 рази, а значення AUCT — в 1,38 рази. Мінімальні остаточні концентрації нінтеданібу залишаються стабільними протягом більше одного року. Виведення Загальний кліренс плазми після внутрішньовенної інфузії є високим (СІ: 1390 мл/хв, gCV — 28.8%). Виведення із сечею незміненої активної речовини впродовж 48 годин після прийому нінтеданібу внутрішньо складає приблизно 0.05% від величини дози (gCV — 31.5%), а після внутрішньовенного введення — приблизно 1.4% (gCV — 24,2%); нирковий кліренс складає 20 мл/хв (gCV — 32,6%). Після прийому внутрішньо [І4С]-нінтеданібу радіоактивний матеріал виводився переважно з жовчю і виявлявся в калі (93.4% дози. gCV — 2.61%). Частка ниркової екскреції в загальному кліренсі є низькою (0.649% дози (gCV — 26,3%)). Виведення вважається повним (більше 90%) через 4 дні після прийому. Період напіввиведення нінтеданібу в термінальній стадії складає від 10 до 15 годин (gCV приблизно 50%).
Лінійність/нелінійність Можна припустити, що фармакокінетика (ФК) нінтеданібу лінійна відносно часу (тобто дані щодо застосування разової дози можуть бути екстрапольовані на дані щодо багаторазового використання). Значення Стах в результаті накопичення препарат) після багаторазового застосування перевищує показник Сщах разової дози в 1,04 рази, а значення AUCT — в 1,38 рази. Мінімальні остаточні концентрації нінтеданібу залишаються стабільними протягом більше одного року. Інша інформація про міжлікарські взаємодії Метаболізм Не очікуються міжлікарські взаємодії між нінтеданібом та субстратами CYP, інгібіторами CYP або індукторами CYP. оскільки нінтеданіб, BIBF 1202 та глюкуронід BIBF 1202 не інгібували або не індукували ферменти CYP під час клінічних досліджень, а також належним чином не метаболізувався ферментами CYP. Транспортування Нінтеданіб є субстратом P—gp. Більш детальна інформація про можливу взаємодію нінтеданібу з цим транспортером наведена в розділі «Взаємодія з іншими лікарськими засобами та інші види взаємодій». Було показано, що нінтеданіб не є субстратом або інгібітором ОАТР-1В1, ОАТР-1ВЗ, ОАТР-2В1, ОСТ-2 або MRP-2 in vitro. Нінтеданіб також не є субстратом BCRP. Спостерігався лише слабкий інгібуючий потенціал на OCT-1. BCRP та P—gp in vitro, що має низьку клінічну значущість, що також може бути застосовано для нінтеданібу як субстрату ОСТ-1. Взаємозв’язок між фармакокінетикою і фармакодинамікою У дослідних фармакокінетичних аналізах побічних реакцій висока експозиція нінтеданібу як правило, була пов’язана з підвищенням рівня печінкових ферментів, а не з побічними реакціями з боку шлунково-кишкового тракту.
Для клінічних кінцевих точок аналіз взаємозв’язку фармакокінетики з ефективністю не проводився. За допомогою методу логістичної регресії встановлений статистично значущий взаємозв’язок між експозицією нінтеданібу та впливом на зміни, які виявляються методом динамічної контрастної магнітно-резонансної томографії. Популяційний фармакокінетичний аналіз v особливих групах пацієнтів Фармакокінетичні властивості нінтеданібу були порівняні у здорових добровольців, пацієнтів з онкологічними захворюваннями та пацієнтів цільової популяції. Стать (скоригована маса тіла), слабке або помірне порушення функції нирок (виміряне шляхом оцінки кліренсу креатиніну). метастази в печінці, бали за шкалою ECOG. вживання алкоголю та генотип P-gp не впливали надію нінтеданібу. Популяційний фармакокінетичний аналіз виявив помірний вплив віку, маси тіла та раси (див. нижче) на дію нінтеданібу. Через високу міжіндивідуальну варіабельність експозиції, що спостерігалась під час клінічного дослідження LUME-Lung-І, ці впливи не вважалися клінічно значущими.
Однак рекомендується проводити ретельне спостереження за станом пацієнтів з декількома з цих факторів ризику (див. розділ «Особливості застосування»). Вік. Експозиція нінтеданібу лінійно збільшується з віком. У 45-річних пацієнтів (5-й процентиль) значення AUCT.SS було нижче на 16%. а у 76-річних пацієнтів (95-й процентиль) вище на 13% в порівнянні з пацієнтами, медіана віку яких складала 62 роки. Діапазон віку, що оцінювався в ході аналізу, складав 29-85 років; вік більше 75 років відзначався приблизно у 5% популяції пацієнтів. Дослідження у дітей не проводилися. Маса тіла. Спостерігається зворотна кореляція між масою тіла і експозицією нінтеданібу. У пацієнтів з масою тіла 50 кг (5-й процентиль) величина AUCT.SS збільшувалася на 25%, а у пацієнтів з масою тіла 100 кг (95-й процентиль) зменшувалася на 19% у порівнянні з пацієнтами, медіана маси тіла яких складала 71.5 кг. Раса. Середня експозиція нінтеданібу на 33 — 50 % вища у пацієнтів-китайців. тайванців та індусів, і на 16 % вище у пацієнтів японців, тоді як у пацієнтів-корейців — на 16 — 22% нижча, ніж у пацієнтів європеоїдної раси (з поправкою на масу тіла). Через високу міжіндивідуальну варіабельність експозиції ці ефекти не були статистично значущими. Дані стосовно пацієнтів негроїдної раси є дуже обмеженими; діапазон цих даних схожий на такий у пацієнтів європеоїдної раси. Порушення функції печінки. Під час спеціального дослідження разової дози фази 1 та порівняно зі здоровими пацієнтами експозиція нінтеданібу на підставі показників Стах та AUC була у 2,2 рази вище у добровольців з легким порушенням функції печінки (клас А за шкалою Чайлд-П*ю; 90% ДІ 1.3 — 3,7 для Cnm та 1,2 — 3.8 для AUC, відповідно). У добровольців з помірним порушенням функції печінки (клас В за шкалою Чайлд-П’ю) експозиція була у 7,6 разів вища на підставі показника Стах (90% ДІ 4,4 — 13.2) та у 8.7- разів вища (90% ДІ 5,7 — 13.1) на підставі показника AUC, відповідно, порівняно зі здоровими добровольцями. Пацієнти з тяжким порушенням функції печінки (клас С за шкалою Чайлд- П’ю) не вивчались.
Клінічні характеристики
Варгатеф Показання
ВАРГАТЕФ показаний в комбінації з доцетакселом для лікування пацієнтів з локально розповсюдженим, метастатичним чи локально рецидивуючим недрібноклітинним раком легенів з ознаками аденокарциноми гістологічного типу після хіміотерапії першої лінії у дорослих.
Протипоказання
Підвищена чутливість до нінтеданібу, арахісу чи сої або до будь-якої із допоміжних речовин препарату
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Р-глікопрогеїн (Р-цр) Нінтеданіб є субстратом P-gp (див. розділ «Фармакологічні властивості. Фармакокінетика»). У спеціальному дослідженні взаємодії препаратів встановлено, що спільне застосування з активним інгібітором P-gp кетоконазолом збільшує експозицію нінтеданібу, судячи з величини AUC, в 1.61 разу, а за показником Стах в 1.83 рази. Спеціальне дослідження взаємодії препаратів продемонструвало, що одночасне застосування рифампіцину (активного індуктора P-gp) призводить до зменшення експозиції нінтеданібу, за показником AUC на 50.3%, а за показником Cmax на 60.3% в порівнянні із застосуванням одного нінтеданібу.
Активні інгібітори P-gp (наприклад кетоконазол або еритроміцин) у разі спільного застосування з нінтеданібом можуть збільшувати експозицію останнього. У таких пацієнтів переносимість нінтеданібу потрібно ретельно відслідковувати. При виникненні побічних реакцій може бути потрібне призупинення терапії, зниження дози або відміна лікування препаратом ВАРГАТЕФ (див. розділ «Спосіб застосування та дози»). Активні індуктори P-gp (наприклад рифампіцин. карбамазепін. фенітоїн і препарати звіробою звичайного) можуть зменшувати експозицію нінтеданібу. Необхідно ретельно зважити супутнє застосування із нінтеданібом. Ізофермент цитохрому (CYP) Ізоферменти CYP беруть лише незначну участь у біотрансформації нінтеданібу.
У доклінічних дослідженнях нінтеданіб та його метаболіти (BIBF 1202 — вільний кислий метаболіт нінтеданібу і його глюкуронід BIBF 1202) не інгібували і не індукували ізоферменти CYP (див. розділ «Фармакологічні властивості. Фармакокінетика»), Тому вірогідність лікарських взаємодій з нінтеданібом, що грунтуються на метаболізмі CYP, вважається невеликою. Одночасне застосування з іншими препаратами Одночасне застосування нінтеданібу з доцетакселом (75 мг/м’) не змінює фармакокінетику жодного з цих речовин у клінічно значимій мірі. Можливість взаємодій нінтеданібу з гормональними контрацептивними засобами не вивчалася.
Особливості застосування
Порушення з боку шлунково-кишкового тракту Діарея була найбільш частим побічним явищем з боку шлунково-кишкового тракту; також відмічається тісний тимчасовий зв’язок між розвитком діареї і застосуванням доцетакселу (див. розділ «Побічні реакції»). Під час клінічного дослідження LUME-Lung 1 (див. розділ «Фармакологічні властивості. Фармакодинаміка») у більшості пацієнтів діарея була легкого і середнього ступеня тяжкості. У післяреєстраційний період були відмічені серйозні випадки діареї, які призвели до дегідратації та порушень електролітного балансу. Діарею слід лікувати при перших ознаках шляхом належної гідратації та протидіарейними засобами, наприклад, лоперамідом. Вона може призвести до призупинення прийому препарату, зниження дози та відміни лікування препаратом ВАРГАТЕФ (див. розділ «Спосіб застосування та дози»). Нудота і блювання, переважно слабкого або помірного ступеня тяжкості, були найчастішими побічними реакціями з боку шлунково-кишкового тракту, про які часто повідомлялося (див. розділ «Побічні реакції»).
Призупинення прийому, зниження дози або припинення лікування препаратом ВАРГАТЕФ (див. розділ «Спосіб застосування та дози») може бути необхідним, незважаючи на відповідну підтримуючу терапію. Підтримуюча терапія нудоти та блювання може включати лікарські засоби з антиеметичною дією, наприклад, глюкокортикоїди, антигістамінні препарати або антагоністи 5НТЗ-рецепторів та адекватну гідратацію. У разі зневоднення слід застосовувати електроліти та рідину. У разі виникнення відповідних побічних явищ з боку шлунково-кишкового тракту слід контролювати рівні електролітів в плазмі крові (див. розділ «Спосіб застосування та дози»). Може бути необхідним призупинення прийому, зниження дози або припинення лікування препаратом ВАРГАТЕФ (див. розділ 4.2).
Нейтропенія та сепсис Висока частота розвитку нейтропенії за критеріями СТСАЕ >3 спостерігалась у пацієнтів, які приймали препарат ВАРГАТЕФ в комбінації з доцетакселом порівняно з монотерапією доцетакселом. Спостерігались такі ускладнення, як сепсис або фебрильна нейтропенія. Під час терапії слід контролювати показники крові, особливо при застосуванні одночасно з доцетакселом. Слід регулярно проводити повний аналіз крові на початку кожного циклу лікування та при максимальному зниженні рівня нейтрофілів, обумовленому проведенням хіміотерапії, пацієнтам, які застосовують нінтеданіб в комбінації з доцетакселом. та за клінічними показаннями після останнього циклу прийому комбінації.
Порушення функції печінки З урахуванням підвищеної експозиції ризик розвитку побічних реакцій може бути підвищений у пацієнтів із слабким порушенням функції печінки (клас А за шкалою Чайлда- П’ю: див. розділи «Спосіб застосування та дози» та «Фармакологічні властивості. Фармакокінетика»). Наявні обмежені дані з безпеки по 9 пацієнтів з гепатоцелюлярною карциномою та помірним порушенням функції печінки (клас В за шкалою Чайлда-П’ю). Незважаючи на відсутність несподіваних висновків щодо безпеки у цих пацієнтів, дані є недостатніми для підтримання рекомендації щодо лікування пацієнтів з помірним порушенням функції печінки. Ефективність нінтеданібу не вивчалась у пацієнтів з помірним порушенням функції печінки (клас В за шкалою Чайлда-ГТю). Безпечність, ефективність та фармакокінетика нінтеданібу не вивчались у пацієнтів з тяжким порушенням функції печінки (Клас С за шкалою Чайлда-П’ю).
Лікування препаратом ВАРГАТЕФ не рекомендується пацієнтам з помірним або тяжким порушенням функції печінки (див. розділ «Спосіб застосу вання та дози»). Прийом нінтеданібу супроводжувався підвищенням рівня ферментів печінки (АЛТ. АСТ, лужної фосфатази (ЛФ), гамма-глютамілтрансферази (ГГТ)). білірубіну та медикаментозним ураженням печінки. У більшості випадків ці підвищення були в основному оборотними при зменшенні дози або призупиненні лікування. Пацієнти з низькою вагою тіла (<65 кг). жіночого роду та пацієнти азіатського походження мають підвищений ризик підвищення рівня ферментів печінки. Експозиція нінтеданібу збільшується лінійно з віком пацієнта, що також може призвести до більш високого ризику збільшення рівня ферментів печінки (див. Розділ «Фармакологічні властивості. Фармакокінетика»). Для пацієнтів з цими факторами ризику рекомендується проводити ретельний моніторинг. Рівні трансфераз, ЛФ та білірубіну слід дослідити до початку комбінованого лікування препаратом ВАРГАТЕФ та доцетакселом. Слід контролювати ці значення за клінічними показаннями або періодично під час лікування, а саме, у фазі комбінованого лікування доцетакселом на початку кожного циклу лікування та щомісяця, якщо продовжують лікування препаратом ВАРГАТЕФ у вигляді монотерапії після припинення лікування доцетакселом.
У разі встановлення відповідного підвищення рівня ферментів печінки, може бути необхідним призупинення прийому, зниження дози або припинення терапії із застосуванням препарату ВАРГАТЕФ (див. розділ «Спосіб застосування та дози»). Слід вивчити альтернативні причини підвищення рівня ферментів печінки та при необхідності, вжити необхідних дій. У разі змін будь-яких показників функції печінки (АСТ/АЛТ > З ВМН (верхня межа норми); загальний білірубін >2 ВМН та ЛФ <2 ВМН). лікування препаратом ВАРГАТЕФ слід припинити. Якщо альтернативна причина не встановлена, лікування препаратом ВАРГАТЕФ слід повністю припинити (див. розділ «Спосіб застосування та дози»). Кровотечі Пригнічення рецептора судинного ендотеліального фактора росту (VEGFR) може бути пов’язано із підвищеним ризиком кровотечі. Під час клінічного випробування (LUME-Lung 1; див. розділ 5.1) препарату ВАРГАТЕФ частота розвитку кровотеч в обох групах лікування була співставлена (див. розділ «Побічні реакції»). Легкі або помірні носові кровотечі були найбільш частим небажаним явищем. Більшість кровотеч з летальним наслідком були пов’язані із пухлинами. Відсутні різниці між респіраторними або фатальними кровотечами та не було повідомлень про внутрішньоцеребральні кровотечі. Пацієнти з нещодавньою легеневою кровотечою (>2,5 мл крові), а також пацієнти з пухлинами центральної локалізації з радіографічним доказом місцевої інвазії головних кровоносних судин або радіографічним доказом наявності кавернозних пухлин або некрозом пухлин були виключені з клінічних досліджень.
Отже, препарат ВАРГАТЕФ не рекомендується застосовувати цим пацієнтам. У післяреєстраційний період були зареєстровані несерйозні та серйозні випадки кровотечі, деякі з яких були смертельними, у тому числі у пацієнтів із антикоагулянтною терапією або без неї, або які лікуватися іншими препаратами, які могли спричинити кровотечу (інформацію стосовно клінічних даних див. «Терапевтична антикоагуляція»), У разі кровотечі, слід враховувати коригування дози, призупинення або припинення лікування, виходячи з клінічної оцінки (див. Розділ 4.2).
Випадки кровотеч, які спостерігатися в післяреєстраційний період включають, але не обмежуються кровотечами з боку органів шлунково-кишкового тракту, дихальної системи та центратьної нервової системи, найчастіше з боку дихальної системи. Терапевтичнії антикоагуляиія Наразі немає даних про пацієнтів зі спадковою схильністю до кровотеч або пацієнтів, які одержувати антикоагулянтну терапію у високих дозах до початку лікування препаратом ВАРГАТЕФ. У пацієнтів, які отримували триваїу терапію низькомолекулярними гепаринами або ацетилсаліциловою кислотою у низьких дозах, не спостерігалося підвищення частоти виникнення кровотеч.
Пацієнтам, у яких розвивались тромбоемболічні події на фоні лікування препаратом та які потребували прийому антикоагулянтів, було дозволено продовжити прийом препарату ВАРГАТЕФ. Не спостерігалося підвищення частоти виникнення кровотеч. Слід регулярно спостерігати за станом пацієнтів, які одночасно застосовували антикоагулянти, такі як варфарин або фенпрокоумон. щодо змін показника протромбінового часу, міжнародного нормалізованого відношення (МНВ) та клінічних епізодів кровотеч. Метастази в головному мозку Стабільні метастази в головному мозку Не спостерігалось підвищення частоти внутрішньомозкових кровотеч у пацієнтів, які отримали належне попереднє лікування метастазів в головному мозку, що були стабільними протягом > 4 тижнів до початку лікування препаратом ВАРГАТЕФ.
Однак стан таких пацієнтів слід ретельно контролювати на наявність ознак та симптомів внутрішньомозкової кровотечі. Активні метастази в головному мозку Пацієнти з активними метастазами в головному мозку були виключені з клінічних досліджень. Цим пацієнтам не рекомендовано застосовувати препарат ВАРГАТЕФ. Венозна тромбоемболія Пацієнти, які застосовують препарат ВАРГАТЕФ. мають підвищений ризик розвитку венозної тромбоемболії, включаючи тромбоз глибоких вен. Стан пацієнтів слід ретельно контролювати на випадок розвитку тромбоемболічних подій. Лікування препаратом ВАРГАТЕФ слід припинити пацієнтам з небезпечною для життя венозною тромбоемболією.
Артеріальні тромбоемболічні події Частота розвитку артеріачьних тромбоемболічних події була порівнювальна між двома групами лікування під час дослідження 1199.13 (ШМЕ-Еипу 1) фази 3. Пацієнти з інфарктом міокарда або інсультом в анамнезі були виключені з цього дослідження. Однак підвищена частота розвитку артеріальних тромбоемболічних події спостерігалась у пацієнтів з ідіопатичним легеневим фіброзом (ІЛФ), які застосовувати нінтеданіб у вигляді монотерапії. Необхідно дотримуватися обережності при лікуванні пацієнтів з високим серцево-судинним ризиком, включаючи відоме захворювання коронарних артерій. Слід розглянути можливість перерви в лікуванні пацієнтів, у яких розвинулися симптоми гострої міокардіаіьної ішемії.
Перфорації шлунково-кишкового тракту Частота розвитку перфорацій шлунково-кишкового тракту була порівнювальна між групами лікування під час клінічного дослідження. Проте у зв’язку з особливостями механізму дії нінтеданібу у пацієнтів, які застосовували препарат ВАРГАТЕФ. може відзначатися підвищення ризику розвитку перфорацій шлунково-кишкового тракту. Особливу увагу слід приділяти лікуванню пацієнтів, які раніше піддавалися абдомінальним хірургічним втручанням або мали перфорації полих органів в анамнезі. У зв’язку з цим. препарат ВАРГАТЕФ можна застосовувати лише як мінімум через 4 тижні після обширних хірургічних втручань. Терапію препаратом ВАРГАТЕФ слід припинити пацієнтам, у яких розвиваються перфорації шлунково-кишкового тракту. Порушення загоєння ран З огляду на механізм дії нінтеданібу, ця речовина може негативно впливати на загоєння ран. У дослідженні ШМЕ-Ьиі^ 1 збільшення частоти порушень загоєння ран не спостерігалося. Спеціальних досліджень, в яких вивчався б вплив нінтеданібу на загоєння ран, не проводилося. Тому лікування препаратом ВАРГАТЕФ має починатися або поновлюватися (якщо здійснювалася перерва у зв’язку з хірургічним втручанням) з урахуванням клінічної оцінки про адекватність загоєння рани.
Вплив на інтервал ОТ Жодних ознак подовження інтервалу О’Г при застосуванні нінтеданібу в рамках програми клінічних досліджень не виявлено (див. розділ «Фармакологічні властивості. Фармакодинаміка»). Оскільки відомо, що деякі інші інгібітори тирозинкінази впливають на ОТ, слід з обережністю призначати нінтеданіб пацієнтам, які знаходяться в групі ризику подовження комплексу ОТ. Алергічні реакції Відомо, що продукти лікувального харчування з вмістом сої спричиняють алергічні реакції, у тому числі тяжкий анафілактичний шок. в осіб із алергією на сою. Пацієнти з відомою алергією на арахісовий білок знаходяться в групі ризику розвитку тяжких реакцій на препарати із вмістом сої. Особливі групи пацієнтів Під час дослідження 1199.13 (ЬиМЕ-Еип§ 1) спостерігалась висока частота розвитку серйозних побічних явищ у пацієнтів з масою тіла менше 50 кг. які застосовували нінтеданіб і доцетаксел, порівняно з пацієнтами з масою тіла > 50 кг; однак кількість пацієнтів з масою тіла менше 50 кг була низькою. Отже, слід ретельно контролювати стан пацієнтів з масою тіла < 50 кг.
Застосування у період вагітності або годування груддю
Жінки репродуктивного віку / контрацепція V жінок Нінтеданіб може чинити негативний вплив на плід людини. Жінкам репродуктивного віку, що приймають препарат ВАРГАТЕФ, слід рекомендувати уникати вагітності. їм потрібно порадити використовувати надійні методи контрацепції під час застосування препарату і протягом щонайменше 3 місяців після прийому останньої дози препарату Варгатеф. Оскільки дія нінтеданібу на метаболізм та ефективність гормональних контрацептивів не вивчалася, другим методом запобігання вагітності повинен стати бар’єрний метод.
Вагітність Спеціальних досліджень щодо застосування препарату ВАРГАТЕФ під час вагітності у людини не проводилося, проте в доклінічних дослідженнях у тварин встановлена репродуктивна токсичність цієї активної речовини. Оскільки нінтеданіб може мати ембріотоксичну дію у людини, його не слід застосовувати під час вагітності, якщо тільки клінічний стан не вимагає проведення лікування. Перед тим. як розпочати лікування препаратом ВАРГАТЕФ. слід провести тест на вагітність. Пацієнткам слід негайно повідомити лікаря про розвиток вагітності під час терапії препаратом ВАРГАТЕФ. Якщо під час терапії препаратом ВАРГАТЕФ розвивається вагітність, пацієнтку необхідно проінформувати про потенційну небезпеку ембріотоксичиої дії препарату. Також слід розглянути питання про припинення лікування препаратом ВАРГАТЕФ.
Годування груддю Відсутні дані про виділення нінтеданібу і його метаболітів в грудне молоко людини. У доклінічних дослідженнях показано, що у тварин в період лактації в грудне молоко проникає невелика кількість нінтеданібу та його метаболітів (< 0.5% від величини дози, що застосовувалася). Тому не можна виключити ризик для новонароджених і грудних дітей. Під час лікування препаратом ВАРГАТЕФ годування груддю слід припинити. Фертильність У доклінічних дослідженнях ознак порушень фертильності у самців виявлено не було. Відсутні дані щодо людей і тварин про потенційний вплив нінтеданібу на жіночу фертильність.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами
ВАРГАТЕФ мас незначний вплив на здатність керувати транспортними засобами та працювати з механізмами. Під час застосування препарату ВАРГАТЕФ пацієнтам потрібно рекомендувати дотримуватися обережності при управлінні транспортними засобами або іншими механізмами.
Спосіб застосування Варгатеф та дози
Лікування препаратом ВАРГАТЕФ слід розпочинати та проводити під наглядом лікаря, який має досвід проведення протипухлинної терапії.
Дози Рекомендована доза нінтеданібу становить 200 мг двічі на добу, приблизно через кожні 12 годин, з 2 по 21 день 21-денного циклу хіміотерапії доцетакселом в стандартному режимі. Варгатеф не слід застосовувати в той самий день, коли вводиться доцетаксел (= День 1). Якщо будь-яка доза препарату була пропущена, то слід продовжній прийом препарату в початково рекомендованій дозі за розкладом наступного прийому препарату. Індивідуальні добові дози нінтеданібу не мають перевищувати рекомендовану дозу для того, щоб компенсувати пропущену дозу. Не слід перевищувати максимальну рекомендовану добову дозу 400 мг. Пацієнти можуть продовжувати терапію нінтеданібом після припинення прийому доцетакселу до досягнення клінічного ефекту або до розвитку неприйнятної токсичності.
Дози, способи застосування та зміни дози доцетакселу наведені у відповідній інструкції для медичного застосування для доцетакселу. Коригування дози Як початковий захід для контролю за побічними реакціями (див. Таблиці 2 та 3), слід тимчасово припинити застосування нінтеданібу до зменшення специфічної побічної реакції до рівня, який би дозволив продовжити лікування (до ступеня 1 або початкового рівня). Лікування нінтеданібом може бути відновлене в пониженій дозі. Коригування дози з кроком у 100 мг на добу (а саме, зниження дози на 50 мг) на підставі індивідуальної безпеки та переносимості рекомендується, як зазначено у Таблиці 2 та Таблиці 3. У разі додаткового розвитку побічної(-их) реакції(-й), а саме, якщо пацієнт не переносить дозу 100 мг двічі на добу, лікування препаратом ВАРГАТЕФ слід зовсім припинити. У разі підвищення рівня аспартатамінотрансферази (ACT)/ аланін-амінотрансферази (АЛТ) більш ніж в 3 рази вище за верхню межу норми одночасно з підвищенням рівня загального білірубіну > 2 рази за верхню межу норми та лужної фосфатази (ЛФ) < 2 рази за верхню межу норми (див. Таблицю 2) лікування препаратом ВАРГАТЕФ слід призупинити. Якщо альтернативна причина не встановлена, застосування препарату ВАРГАТЕФ слід повністю припинити (див. також розділ «Особливості застосування»).
Таблиця 2. Рекомендоване коригування дози препарату ВЛРГАТЕФ (нінтеданіб) у разі розвитку діареї. блювання та інших гематологічних та негематологічних побічних реакцій

* СТСАЕ: Загальна термінологія критеріїв побічних явищ
Таблиця 3. Рекомендоване коригування дози препарату ВАРГАТЕФ (нінтеданіб) у разі підвищення

ACT: аспартатамінотрансфераза; АЛТ: аланін-амінотрансфераза ЛФ: лужна фосфатаза; ВМН: верхня межа норми
Особливі групи пацієнтів
Дитячий вік Безпечність та ефективність застосування препарату ВАРГАТЕФ дітям (віком до 18 років) не встановлені. Дані відсутні. Пацієнти літнього віку (> 65 років) Не відзначено жодних загальних відмінностей з точки зору безпеки та ефективності застосування препарату літнім пацієнтам. Під час проведення базового дослідження 1199.13 85 пацієнтів (12,9 % пацієнтів з гістологічно виявленою аденокарциномою) були у віці > 70 років (середній вік: 72 років, діапазон: 70 — 80 років) (див. розділ «Фармакологічні властивості. Фармакодинаміка»), Коригування початкової дози препарату залежно від віку пацієнта не потрібне, (див. розділ «Фармакологічні властивості. Фармакокінетика»).
Раса та маса тіла На підставі даних популяційного фармакокінетичного аналізу теоретично коригувати дозу препарату ВАРГАТЕФ не потрібно (див. розділ «Фармакологічні властивості. Фармакокінетика»). Дані з безпеки щодо пацієнтів негроїдної та афро-американської раси обмежені. Порушення функції нирок Нирками виводиться менше 1% разової дози нінтеданібу (див. розділ «Фармакологічні властивості. Фармакокінетика»), Пацієнтам з порушеннями функції нирок легкого або помірного ступеня тяжкості коригувати початкову дозу не потрібно. Для пацієнтів з тяжкими порушеннями функції нирок (кліренс креатиніну < 30 мл/хв.) безпека, ефективність та фармакокінетика нінтеданібу не вивчалися.
Порушення функції печінки
Нінтеданіб виводиться переважно з жовчю/калом (> 90%). Дія посилюється у пацієнтів із порушенням функції печінки (клас А та клас В за шкалою Чайлда-ГГю; див. розділ «Фармакологічні властивості. Фармакокінетика»). На підставі даних клінічних досліджень коригу вати початкову дозу не потрібно пацієнтам з порушеннями функції печінки легкого ступеня тяжкості (клас А за шкалою Чайлда-ГГю). Обмежені дані з безпеки по 9 пацієнтам з помірним порушенням функції печінки (клас В за шкалою Чайлда-П’ю) є недостатніми, щоб характеризувати цю популяцію. У пацієнтів з тяжким порушенням функції печінки (клас С за шкалою Чайлда-ГГю) безпека, ефективність та фармакокінетика нінтеданібу не вивчалася. Тому лікування пацієнтів з порушеннями функції печінки помірного (клас В за шкалою Чайлда-ГГю) і тяжкого (клас С за шкалою Чайлда-П’ю) ступеня препаратом ВАРГАТЕФ не рекомендується (див. розділи «Особливості застосування» та «Фармакологічні властивості. Фармакокінетика»).
Діти
Препарат не застосовують у педіатричній практиці.
Передозування
Симптоми
Найвища разова доза нінтеданібу. яка застосовувалась під час проведення досліджень фази І, становила 450 мг один раз на добу. Зафіксовані випадки передозування у двох пацієнтів при застосуванні препарату в максимальній дозі 600 мг двічі на добу впродовж восьми днів. Небажані явища, що спостерігалися, були порівнянні з відомим профілем безпеки нінтеданібу: збільшення активності ферментів печінки і порушення з боку ШКТ. Обидва пацієнти повністю відновилися після небажаних явищ.
Лікування
Специфічного антидоту або способу лікування передозування нінтеданібу немає. У разі передозування необхідно відмінити препарат і проводити симптоматичну терапію.
Побічні реакції
Дані з безпеки, наведені в розділах нижче, базуються на підставі результатів глобального, подвійного сліпого, рандомізованого базового дослідження фази III 1199.13 (LUME—Lung 1), у ході якого порівнювалося лікування нінтеданібом і доцетакселом з курсом лікування плацебо і доцетакселом у пацієнтів з локально розповсюдженим або метастатичним чи рецидивуючим недрібноклітинним раком легенів після хіміотерапії першої лінії. Найбільш частими побічними реакціями на фоні застосування нінтеданібу були діарея, підвищення рівня ферментів печінки (АЛТ та ACT) та блювання. В таблиці 4 підсумовані побічні реакції відповідно до класів систем органів. Для одержання інформації стосовно лікування окремих побічних реакцій див. розділ «Особливості застосування». Інформація про окремі побічні реакції, шо спостерігались під час проведення дослідження LUME—Lung 1. наведена нижче.
Перелік побічних реакцій (ГІР)
У таблиці 4 підсумована частота побічних реакцій, про які повідомлялося під час проведення базового дослідження LUME—Lung 1, у пацієнтів з недрібноклітинним раком легенів з ознаками аденокарциноми гістологічного типу (п = 320). Побічні явища наведені відповідно до частоти виникнення: дуже часті (>1/10), часті (> 1/100 до < 1/10), нечасті (> 1/1,000 до < 1/100), поодинокі (> 1/10,000 до < 1/1,000), рідкісні (< 1/10 000), невідомо (неможливо встановити за наявними даними). У межах кожної групи за частотою небажані реакції представлено в порядку зменшення серйозності.
Таблиця 4. Резюме побічних реакцій на лікарський засіб за категоріями частоти
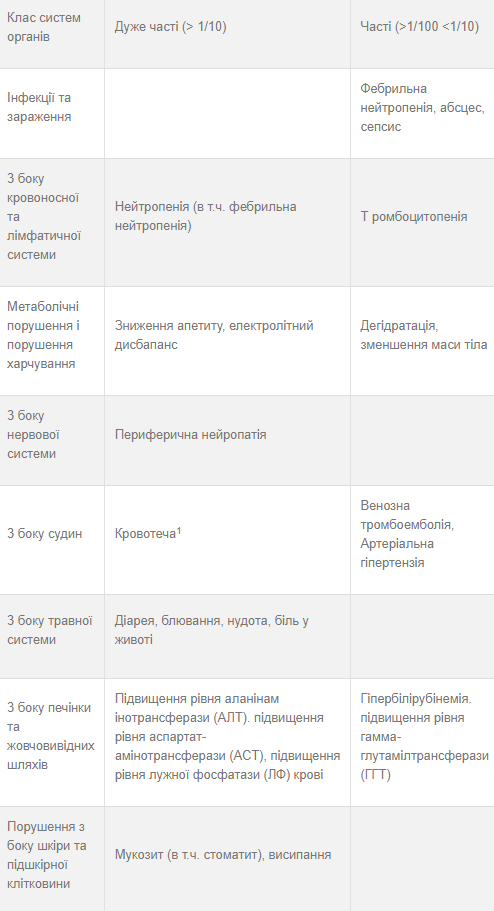

1 Частота розвитку не збільшувалась у пацієнтів, які застосовували нінтеданіб і доцетаксел, порівняно з плацебо і доцетаксел. 2 Випадки панкреатиту спостерігалися у пацієнтів, які приймали нінтеданіб для лікування ідіопатичного легеневого фіброзу та недрібноклітинного раку легенів. Більшість цих випадків спостерігалися у пацієнтів за показанням ідіопатичного легеневого фіброзу.
Опис окремих побічних реакцій
Діарея
Діарея відзначалася у 43,4 % (> ступеня 3: 6,3 %) пацієнтів, з аденокарциномою в групі нінтеданібу. Більшість побічних реакцій була тимчасово тісно пов’язана із прийомом доцетакселу. У більшості пацієнтів прояви діареї зникали після припинення лікування, проведення протидіарейної терапії та зменшення дози нінтеданібу. Рекомендовані заходи та коригування доз у разі діареї див. у розділах «Особливості застосування» та «Спосіб застосування та дози», відповідно.
Підвищення рівнів ферментів печінки та гіпербиіірубінемія
Побічні реакції з боку печінки виникали у 42,8 % пацієнтів, які приймали нінтеданіб. Приблизно у однієї третини цих пацієнтів відмічали побічні реакції з боку печінки > З ступеня тяжкості. У пацієнтів з підвищеними печінковими параметрами застосування встановленої схеми поступового зниження дози було необхідною мірою, а припинення лікування було необхідним тільки у 2,2 % пацієнтів. У більшості пацієнтів підвищення печінкових параметрів були оборотними. Інформацію стосовно особливих груп пацієнтів, рекомендованих заходів та коригування доз у разі підвищення рівня печінкових ферментів та білірубіну можна знайти у розділах «Особливості застосування» та «Спосіб застосування та дози», відповідно.
Нейтропенія.
Фебриіьна нейтропенія та сепсис. Сепсис та фебрильна нейтропенія спостерігатись як ускладнення нейтропенії. Частота розвитку сепсису (1.3 %) та фебрильної нейтропенії (7.5 %) збільшувалась на фоні прийому нінтеданібу порівняно з групою плацебо. Під час терапії важливо проводити аналіз крові пацієнта, особливо під час комбінованого лікування доцетакселом (див. розділ «Особливості застосування»).
Кровотеча
У післяреєстраційному періоді були зареєстровані несерйозні та серйозні кровотечі, деякі з яких були фатальними, включаючи пацієнтів з терапією антикоагулянтами або без них, або іншими препаратами, які можуть спричинити кровотечу. Випадки кровотечі у післіреєстраційному періоді включають, але не обмежуються, з боку органів шлунково- кишкового тракту, органів дихання та центральної нервової системи, причому найчастіше з боку органів дихання (див. також розділ «Особливості застосування»).
Перфорація
Як очікується, у зв’язку з особливостями механізму дії перфорація може виникнути у пацієнтів, які застосовували нінтеданіб. Однак кількість пацієнтів з перфорацією шлунково- кишкового тракту була низькою.
Периферична нейропатія
Також відомо, що периферична нейропатія може виникати на фоні лікування доцетакселом. Периферична нейропатія спостерігалась у 16.5 % пацієнтів в групі плацебо та у 19.1 % пацієнтів в групі нінтеданібу.
Повідомлення про побічні реакції
Повідомлення про побічні реакції після реєстрації лікарського препарату є важливими. Це дає змогу постійно вести моніторинг співвідношення користь/ризик лікарського засобу. Просимо медичних працівників повідомляти про будь-які можливі побічні реакції за допомогою національної системи звітності.
Термін придатності Варгатеф
З роки.
Умови зберігання Варгатеф
Зберігати при температурі не вище 25°С, в оригінальній упаковці для захисту від вологи.
Упаковка
Для дозування 100 мг: по 10 капсул в алюмінієвому блістері, по 6 або 12 блістерів в картонній упаковці.
Категорія відпуску
За рецептом.
Виробник
Берінгер Інгельхайм Фарма ГмбХ і Ко. КГ/Boehringer Ingelheim Pharma GmbH & Co. KG.
Місцезнаходження виробника
Бінгер Штрассе 173, 55216, Інгельхайм на Рейні, Німеччина Binger Strasse 173, 55216, Ingelheim am Rhein, Germany.
Подальша інформація
Пам'ятайте, зберігайте ці та всі інші ліки в недоступному для дітей місці, ніколи не передавайте свої ліки іншим і використовуйте Варгатеф тільки за призначенням.
Завжди консультуйтеся зі своїм лікарем, щоб переконатися, що інформація, яка відображається на цій сторінці, може бути застосована до ваших особистих обставин.
Увага! Ця інструкція для медичного застосування лікарського засобу є офіційною інструкцією виробника Берінгер Інгельхайм Фарма ГмбХ і Ко. КГ.
Авторське право:
- Берінгер Інгельхайм Фарма ГмбХ і Ко. КГ
| Тип даних | Відомості з реєстру |
| Торгівельне найменування: | Варгатеф |
| Виробник: | Берінгер Інгельхайм Фарма ГмбХ і Ко. КГ |
| Форма випуску: | капсули м`які по 100 мг або 150 мг, по 10 капсул м'яких у алюмінієвому блістері, по 6 або 12 блістерів у картонній коробці |
| Реєстраційне посвідчення: | UA/16651/01/01, UA/16651/01/02 |
| Дата початку: | 26.04.2018 |
| Дата закінчення: | 26.04.2023 |
| Міжнародне непатентоване найменування: | Nintedanib |
| Умови відпуску: | за рецептом |
| Склад: | 1 капсула містить: нінтеданібу 100 мг (120,40 мг у вигляді езилату) |
| Фармакотерапевтична група: | Антинеопластичні засоби. Інгібітори протеїнкінази. |
| Код АТС: | L01XE31 |
| Заявник: | Берінгер Інгельхайм Інтернешнл ГмбХ |
| Країна заявника: | Німеччина |
| Адреса заявника: | Бінгер Штрассе 173, D-55216 Інгельхайм-на-Рейні, Нiмеччина |
| Тип ЛЗ: | Звичайний |
| ЛЗ біологічного походження: | Нi |
| ЛЗ рослинного походження: | Нi |
| Гомеопатичний ЛЗ: | Нi |
| Тип МНН: | Моно |
| Дострокове припинення | Нi |
| Код ATC | Назва групи |
| L | Антинеопластичні та імуномодулюючі засоби |
| L01 | Протипухлинні препарати |
| L01X | Інші антинеопластичні засоби |
| L01XE | Інгібітори протеїнкінази |
| L01XE31 | Нінтеданіб |