


- Склад
- Лікарська форма
- Фармакотерапевтична група
- Фармакологічні властивості
- Клінічні характеристики
- Показання
- Протипоказання
- Взаємодія з іншими засобами
- Особливості застосування
- У період вагітності
- При керуванні автомобілем
- Спосіб застосування та дози
- Діти
- Передозування
- Побічні реакції
- Термін придатності
- Умови зберігання
- Упаковка
- Категорія відпуску
- Виробник
- Місцезнаходження виробника
Скафо інструкція із застосування
Офіційна інструкція лікарського засобу Скафо порошок 150 мг. Опис та застосування Skafo, аналоги та відгуки. Інструкція Скафо порошок затверджена виробником.
Склад
діюча речовина:секукінумаб (secukinumab);
1 флакон з порошком містить 150 мг секукінумабу;
після відновлення 1 мл розчину містить 150 мг секукінумабу;
допоміжні речовини: сахароза, L-гістидин, L-гістидину гідрохлориду моногідрат, полісорбат 80.
Лікарська форма
Порошок для розчину для ін’єкцій.
Основні фізико-хімічні властивості: порошок являє собою твердий ліофілізат білого кольору.
Фармакотерапевтична група
Імуносупресанти, інгібітори інтерлейкіну.
Код АТХ L04A C10.
Фармакологічні властивості
Фармакодинаміка.
Механізм дії
Секукінумаб являє собою повністю людське моноклональне антитіло IgG1/κ, що селективно зв’язується та нейтралізує прозапальний цитокін - інтерлейкін‑17A (IL‑17A). Секукінумаб чинить спрямовану дію на IL‑17A та інгібує його взаємодію з рецептором IL‑17, який експресується різними типами клітин, включаючи кератиноцити. В результаті цього секукінумаб уповільнює вивільнення прозапальних цитокінів, хемокінів і медіаторів пошкодження тканин, знижує внесок ІЛ-17A в аутоімунні і запальні захворювання. Секукінумаб в клінічно значущих концентраціях проникає в шкіру і знижує концентрацію запальних маркерів. Прямим наслідком лікування секукінумабом є зменшення вираженості почервоніння, ущільнення і лущення шкіри, що спостерігається в осередках ураження при бляшковому псоріазі.
IL‑17A є природним цитокіном, який бере участь в природних реакціях запалення і імунної відповіді. IL‑17A відіграє ключову роль в патогенезі бляшкового псоріазу, псоріатичного артриту та аксіального спондилоартриту (анкілозуючого спондиліту і нерентгенологічного аксіального спондилоартриту) та регулюється в ураженій шкірі на відміну від такого в неураженій шкірі пацієнтів з бляшковим псоріазом, а також в синовіальній тканині пацієнтів із псоріатичним артритом. У пацієнтів з анкілозуючим спондилітом значне збільшення кількості клітин, що продукують IL-17A, відмічається в субхондральному кістковому мозку фасеткових суглобів. Також в крові у пацієнтів з нерентгенологічним аксіальним спондилоартритом виявлено збільшення концентрації IL-17А. Виявлено, що пригнічення
IL-17A є ефективним при лікуванні анкілозуючого спондиліту, таким чином, встановлено ключову роль цього цитокіну в аксіальному спондилоартриті.
Фармакодинамічні ефекти
Сироваткові рівні загального IL-17A (вільного і зв’язаного з секукінумабом IL-17A) спочатку збільшувалися у пацієнтів, які отримували секукінумаб. Це відбувалося внаслідок зниження кліренсу пов’язаного з секукінумабом IL-17A, що вказує на те, що секукінумаб селективно зв’язується з вільним IL-17A, відіграючи ключову роль у патогенезі бляшкового псоріазу.
Під час дослідження у пацієнтів з бляшковим псоріазом після одного-двох тижнів лікування секукінумабом значно знижувалися інфільтрація епідермісу нейтрофілами і кількість різних асоційованих з ними маркерів, що часто підвищені в уражених ділянках шкіри у таких пацієнтів.
Показано, що секукінумаб знижує (протягом 1-2 тижнів лікування) рівні C-реактивного білка, який є маркером запалення.
Клінічна ефективність та безпека
Бляшковий псоріаз у дорослих
Безпеку та ефективність застосування секукінумабу оцінювали під час чотирьох рандомізованих, подвійно сліпих, плацебо-контрольованих досліджень III фази у пацієнтів з помірним або тяжким бляшковим псоріазом, яким показана фототерапія або системна терапія (ERASURE, FIXTURE, FEATURE, JUNCTURE).
Ефективність і безпеку застосування секукінумабу в дозі 150 мг та 300 мг оцінювали порівняно з плацебо або етанерцептом. Крім того, в одному дослідженні оцінювався режим тривалої терапії порівняно із застосуванням у разі потреби (SCULPTURE).
З 2403 пацієнтів, включених у плацебо-контрольовані дослідження, 79 % не отримували біологічних лікарських засобів, 45 % не відповіли на терапію небіологічними препаратами, а 8 % не відповіли на терапію біологічними препаратами (6 % не відповіли на терапію анти-ФНП і 2 % не відповіли на терапію анти-р40). Під час досліджень III фази приблизно у
15-25 % пацієнтів відмічений псоріатичний артрит (ПА) на початку дослідження.
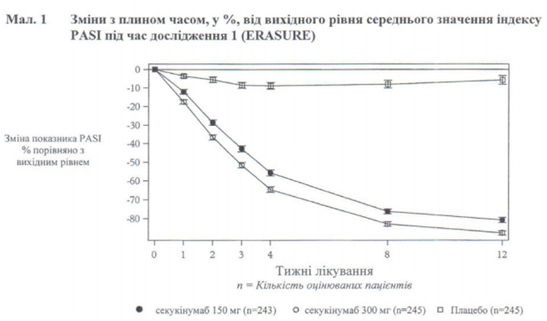
В дослідженні 1 псоріазу (ERASURE) оцінювали 738 пацієнтів. Пацієнти, які були рандомізовані до групи секукінумабу, отримували дози 150 мг або 300 мг на 0, 1, 2 і 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4-го тижня. В дослідженні 2 псоріазу (FIXTURE) оцінювали 1 306 пацієнтів. Пацієнти, які були рандомізовані до групи секукінумабу, отримували дози 150 мг або 300 мг на 0, 1, 2 і 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4-го тижня. Пацієнти, рандомізовані до групи етанерцепту, отримували дозу 50 мг двічі на тиждень протягом 12 тижнів, потім 50 мг щотижня. В обох дослідженнях (1 і 2) пацієнти, які були рандомізовані до групи плацебо і які не відповіли на лікування до 12-го тижня, перейшли на секукінумаб (150 мг або 300 мг) на 12, 13, 14 і 15 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 16-го тижня. Всі пацієнти проходили подальше спостереження до 52 тижнів після початку лікування.
В дослідженні псоріазу 3 (FEATURE) оцінювали 177 пацієнтів, які застосовували секукінумаб у попередньо наповненому шприці порівняно з плацебо через 12 тижнів після початку лікування для оцінки безпеки, переносимості та користі самостійного застосування секукінумабу у попередньо наповненому шприці. В дослідженні псоріазу 4 (JUNCTURE) оцінювали 182 пацієнтів, які застосовували секукінумаб у попередньо наповненій шприц-ручці порівняно з плацебо, через 12 тижнів після початку лікування для оцінки безпеки, переносимості та користі самостійного застосування секукінумабу у попередньо наповненій шприці-ручці. В обох дослідженнях (3 та 4) пацієнти були рандомізовані до групи секукінумабу в дозах 150 мг або 300 мг на 0, 1, 2 і 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4-го тижня. Пацієнти були також рандомізовані до групи плацебо на 0, 1, 2 і 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4-го тижня.
В дослідженні псоріазу 5 (SCULPTURE) оцінювали 966 пацієнтів. Всі пацієнти отримували секукінумаб в дозах 150 мг або 300 мг на 0, 1, 2, 3, 4, 8 та 12 тижні та потім були рандомізовані до групи щомісячного введення підтримуючої дози, починаючи з 12-го тижня, або до групи лікування у разі потреби в тій самій дозі. Пацієнти, рандомізовані до режиму лікування у разі потреби, не досягли задовільної відповіді на терапевтичне лікування, а отже, рекомендується щомісячна підтримуюча терапія фіксованими дозами.
Первинними складовими кінцевих точок в плацебо-контрольованих дослідженнях і дослідженнях з активним контролем були відсотки пацієнтів, які досягли PASI 75 і значень ІGA mod 2011 «чисто» або «майже чисто» порівняно з плацебо на 12-му тижні (див. таблиці 1 і 2). Доза 300 мг збільшує шкірний кліренс практично до значень «чисто» або «майже чисто» в кінцевих точках ефективності PASI 90, PASI 100 та IGA mod 2011 відповідно до індексу 0 або 1 у всіх дослідженнях з досягненням піка на 16-му тижні, отже, ця доза є рекомендованою.
Таблиця 1 Короткий огляд PASI 50/75/90/100 та IGA⃰ mod 2011 «чисто» або «майже чисто» клінічної відповіді в рамках досліджень 1, 3 та 4 псоріазу (ERASURE, FEATURE та JUNCTURE)
Показники | Тиждень 12 | Тиждень 16 | Тиждень 52 | ||||||
плацебо | секукінумаб | секукінумаб | секукінумаб | ||||||
150 мг | 300 мг | 150 мг | 300 мг | 150 мг | 300 мг | ||||
Дослідження 1 | |||||||||
Кількість пацієнтів | 246 | 244 | 245 | 244 | 245 | 244 | 245 | ||
Відповідь PASI 50, n (%) | 22 (8,9 %) | 203 (83,5 %) | 222 (90,6 %) | 212 (87,2 %) | 224 (91,4 %) | 187 (77 %) | 207 (84,5 %) | ||
Відповідь PASI 75, n (%) | 11 (4,5 %) | 174 (71,6%)** | 200 (81,6 %)** | 188 (77,4 %) | 211 (86,1 %) | 146 (60,1 %) | 182 (74,3 %) | ||
Відповідь PASI 90, n (%) | 3 (1,2 %) | 95 (39,1%)** | 145 (59,2 %)** | 130 (53,5 %) | 171 (69,8 %) | 88 (36,2 %) | 147 (60,0 %) | ||
Відповідь PASI 100, n (%) | 2 (0,8 %) | 31 (12,8 %) | 70 (28,6 %) | 51 (21,0 %) | 102 (41,6 %) | 49 (20,2 %) | 96 (39,2 %) | ||
Відповідь IGA mod 2011 «чисто» або «майже чисто», n (%) | 6 (2,40 %) | 125 (51,2%)** | 160 (65,3 %)** | 142 (58,2 %) | 180 (73,5 %) | 101 (41,4 %) | 148 (60,4 %) | ||
Дослідження 3 | |||||||||
Кількість пацієнтів | 59 | 59 | 58 | ‑ | ‑ | ‑ | ‑ | ||
Відповідь PASI 50, n (%) | 3 (5,1 %) | 51 (86,4%) | 51 (87,9 %) | ‑ | ‑ | ‑ | ‑ | ||
Відповідь PASI 75, n (%) | 0 (0,0 %) | 41 (69,5%)** | 44 (75,9 %)** | ‑ | ‑ | ‑ | ‑ | ||
Відповідь PASI 90, n (%) | 0 (0,0 %) | 27 (45,8 %) | 35 (60,3 %) | ‑ | ‑ | ‑ | ‑ | ||
Відповідь PASI 100, n (%) | 0 (0,0 %) | 5 (8,5 %) | 25 (43,1 %) | ‑ | ‑ | ‑ | ‑ | ||
Відповідь IGA mod 2011 «чисто» або «майже чисто», n (%) | 0 (0,0 %) | 31 (52,5%)** | 40 (69,0 %)** | ‑ | ‑ | ‑ | ‑ | ||
Дослідження 4 | |||||||||
Кількість пацієнтів | 61 | 60 | 60 | ‑ | ‑ | ‑ | ‑ | ||
Відповідь PASI 50, n (%) | 5 (8,2 %) | 48 (80,0 %) | 58 (96,7 %) | ‑ | ‑ | ‑ | ‑ | ||
Відповідь PASI 75, n (%) | 2 (3,3 %) | 43 (71,7%)** | 52 (86,7 %)** | ‑ | ‑ | ‑ | ‑ | ||
Відповідь PASI 90, n (%) | 0 (0,0 %) | 24 (40,0 %) | 33 (55,0 %) | ‑ | ‑ | ‑ | ‑ | ||
Відповідь PASI 100, n (%) | 0 (0,0 %) | 10 (16,7 %) | 16 (26,7 %) | ‑ | ‑ | ‑ | ‑ | ||
Відповідь IGA mod 2011 «чисто» або «майже чисто», n (%) | 0 (0,0 %) | 32 (53,3%)** | 44 (73,3 %)** | ‑ | ‑ | ‑ | ‑ | ||
*IGA mod 2011 - це 5-бальна шкала, включаючи індекси 0 - «чисто», 1 - «майже чисто», 2 - «легкий ступінь», 3 - «помірний ступінь» або 4 - «тяжкий ступінь», що свідчить про загальну лікарську оцінку тяжкості псоріазу з огляду на затвердіння, еритему і лущення шкіри. Ефект лікування, який визначається як «чисто» або «майже чисто», полягає у відсутності ознак псоріазу або уражень природного або рожевого кольору, відсутності потовщення бляшок, а також у відсутності або мінімальному місцевому лущенні шкіри.
**Значення р проти плацебо і скориговані з урахуванням кратності: p < 0,0001.
Таблиця 2 Короткий огляд клінічної відповіді в рамках дослідження 2 псоріазу (FIXTURE)
Показники | Тиждень 12 | Тиждень 16 | Тиждень 52 | |||||||
плацебо | секукінумаб | етанер-цепт | секукінумаб | етанерцепт | секукінумаб | етанер-цепт | ||||
150 мг | 300 мг | 150 мг | 300 мг | 150 мг | 300 мг | |||||
Кількість пацієнтів | 324 | 327 | 323 | 323 | 327 | 323 | 323 | 327 | 323 | 323 |
Відповідь PASI 50, n (%) | 49 (15,1 %) | 266 (81,3 %) | 296 (91,6 %) | 226 (70,0 %) | 290 (88,7 %) | 302 (93,5 %) | 257 (79,6 %) | 249 (76,1 %) | 274 (84,8 %) | 234 (72,4 %) |
Відповідь PASI 75, n (%) | 16 (4,9 %) | 219 (67,0%)** | 249 (77,1%)** | 142 (44,0 %) | 247 (75,5 %) | 280 (86,7 %) | 189 (58,5 %) | 215 (65,7 %) | 254 (78,6 %) | 179 (55,4 %) |
Відповідь PASI 90, n (%) | 5 (1,5 %) | 137 (41,9 %) | 175 (54,2 %) | 67 (20,7 %) | 176 (53,8 %) | 234 (72,4 %) | 101 (31,3 %) | 147 (45,0 %) | 210 (65,0 %) | 108 (33,4 %) |
Відповідь PASI 100, n (%) | 0 (0 %) | 47 (14,4 %) | 78 (24,1 %) | 14 (4,3 %) | 84 (25,7 %) | 119 (36,8 %) | 24 (7,4 %) | 65 (19,9 %) | 117 (36,2 %) | 32 (9,9 %) |
Відповідь IGA mod 2011 «чисто» або «майже чисто», n (%) | 9 (2,8 %) | 167 (51,1%)** | 202 (62,5%)** | 88 (27,2 %) | 200 (61,2 %) | 244 (75,5 %) | 127 (39,3 %) | 168 (51,4 %) | 219 (67,8 %) | 120 (37,2 %) |
**p значення проти етанерцепту: p = 0,0250.
У додатковому дослідженні псоріазу (CLEAR) оцінювали 676 пацієнтів. Секукінумаб в дозі 300 мг відповідав первинній та вторинній кінцевим точкам, демонструючи більшу ефективність, ніж устекінумаб, на підставі відповіді PASI 90 на 16-му тижні (первинна кінцева точка), швидкості початку відповіді PASI 75 на 4-му тижні та тривалої відповіді PASI 90 на 52-му тижні. Більш висока ефективність у порівнянні з устекінумабом для кінцевих точок PASI 75/90/100 та IGA mod 2011 відповідно до індексу 0 або 1 («чисто» або «майже чисто») спостерігалася на початку та продовжувалася до 52-го тижня.
Таблиця 3 Короткий огляд клінічної відповіді в рамках дослідження CLEAR
Показники | Тиждень 4 | Тиждень 16 | Тиждень 52 | |||
Секукінумаб 300 мг | Устекінумаб* | Секукінумаб 300 мг | Устекінумаб* | Секукінумаб 300 мг | Устекінумаб* | |
Кількість пацієнтів | 334 | 335 | 334 | 335 | 334 | 335 |
Відповідь PASI 75, n (%) | 166 (49,7 %)** | 69 (20,6 %) | 311 (93,1 %) | 276 (82,4 %) | 306 (91,6 %) | 262 (78,2 %) |
Відповідь PASI 90, n (%) | 70 (21,0 %) | 18 (5,4 %) | 264 (79,0 %)** | 192 (57,3 %) | 250 (74,9 %)*** | 203 (60,6 %) |
Відповідь PASI 100, n (%) | 14 (4,2 %) | 3 (0,9 %) | 148 (44,3 %) | 95 (28,4 %) | 150 (44,9 %) | 123 (36,7 %) |
Відповідь IGA mod 2011 «чисто» або «майже чисто», n (%) | 128 (38,3 %) | 41 (12,2 %) | 278 (83,2 %) | 226 (67,5 %) | 261 (78,1 %) | 213 (63,6 %) |
*Пацієнти, які отримували секукінумаб, приймали дозу 300 мг на 0, 1, 2, 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4 тижня до 52 тижня. Пацієнти, які отримували устекінумаб, приймали 45 мг або 90 мг на 0 та 4-ому тижні, потім кожні 12 тижнів до 52 тижня (дозування з урахуванням маси тіла відповідно до схваленої дози).
**p значення проти устекінумабу: p < 0,0001 для первинної кінцевої точки PASI 90 на 16-ому тижні та вторинної кінцевої точки PASI 75 на 4-ому тижні.
***p значення проти устекінумабу: p = 0,0001 для вторинної кінцевої точки PASI 90 на
52-ому тижні.
Секукінумаб продемонстрував ефективність у пацієнтів, які раніше не отримували системну терапію та терапію біологічними препаратами, у пацієнтів, які отримували терапію біологічними препаратами/анти-ФНП, а також у пацієнтів з недостатньою відповіддю на терапію біологічними препаратами/анти-ФНП. Покращення індексу PASI 75 у пацієнтів із супутнім псоріатичним артритом на початку дослідження аналогічне такому у загальній популяції пацієнтів із бляшковим псоріазом.
Прийом секукінумабу в дозі 300 мг був пов’язаний зі швидким початком прояву ефективності зі зниженням середнього показника PASI на 50 % на 3 тижні.
Специфічна локалізація/ форми бляшкового псоріазу
Під час двох додаткових плацебо-контрольованих досліджень спостерігалося зменшення симптомів псоріазу нігтів (TRANSFIGURE, 198 пацієнтів) та долонно-підошовного бляшкового псоріазу (GESTURE, 205 пацієнтів). Під час дослідження TRANSFIGURE секукінумаб був більш ефективним, ніж плацебо, на 16-ому тижні (46,1 % в групі прийому 300 мг, 38,4 % в групі прийому 150 мг та 11,7 % в групі прийому плацебо) з точки зору значущого покращення відносно вихідних показників відповідно до індексу тяжкості псоріатичного ураження нігтів (NAPSI %) у пацієнтів з помірним або тяжким бляшковим псоріазом з ураженням нігтів. Під час дослідження GESTURE секукінумаб був більш ефективним, ніж плацебо, на 16-ому тижні (33,3 % в групі прийому 300 мг, 22,1 % в групі прийому 150 мг та 1,5 % в групі прийому плацебо) з точки зору значущого покращення ppIGA значень 0 або 1 («чисто» або «майже чисто») у пацієнтів з помірним або тяжким долонно-підошовним бляшковим псоріазом.
Під час плацебо-контрольованого дослідження оцінювали 102 пацієнтів з помірним або тяжким псоріазом шкіри голови з індексом тяжкості псоріазу шкіри голови (PSSI) ≥ 12, індексом IGA mod 2011 шкіри голови 3 або більше та щонайменше 30 % загальної площі ураженої шкіри голови. Секукінумаб в дозі 300 мг був більш ефективним, ніж плацебо, на
12-ому тижні з точки зору значущого покращення відносно вихідних показників відповідно до показника PSSI 90 (52,9 % проти 2,0 %) та показника IGA mod 2011 0 або 1 шкіри голови (56,9 % проти 5,9 %). Покращення за кожною кінцевою точкою зберігалися у пацієнтів, які продовжили приймати секукінумаб до 24-го тижня.
Якість життя/ результати, відмічені пацієнтами
Статистично значущі покращення на 12-ому тижні (дослідження 1-4) відносно вихідних показників порівняно з плацебо були продемонстровані відповідно до DLQI (дерматологічний індекс якості життя). Середні зниження (покращення) відповідно до DLQI відносно вихідних показників варіювали від ‑10,4 до ‑11,6 у пацієнтів, які приймали секукінумаб в дозі 300 мг, від ‑7,7 до ‑10,1 у пацієнтів, які приймали секукінумаб в дозі 150 мг, проти ‑1,1 - ‑1,9 у пацієнтів, які приймали плацебо на 12-ому тижні. Такі покращення зберігались протягом 52 тижнів (дослідження 1 та 2).
Сорок відсотків учасників досліджень 1 та 2 заповнили Щоденник Симптомів Псоріазу©. Учасники, які заповнювали щоденник в кожному з цих досліджень, показали статистично значущі покращення на 12-ому тижні відносно вихідних показників порівняно з плацебо щодо ознак та симптомів свербежу, болю та лущення шкіри.
Статистично значущі покращення на 4-ому тижні відносно вихідних показників у пацієнтів, які отримували секукінумаб, порівняно з пацієнтами, які отримували устекінумаб (CLEAR), були продемонстровані в DLQI, та ці покращення зберігалися протягом 52 тижнів.
Статистично значущі покращення у пацієнтів, у яких відмічались ознаки та симптоми свербежу, болю та лущення шкіри голови на 16 та 52 тижні (CLEAR), були продемонстровані у Щоденнику Симптомів Псоріазу© у пацієнтів, які отримували секукінумаб, порівняно з пацієнтами, які отримували устекінумаб.
У дослідженні псоріазу шкіри голови були продемонстровані статистично значущі покращення (зменшення) на 12-ому тижні відносно вихідних показників у пацієнтів, у яких відмічались ознаки та симптоми свербежу, болю та лущення шкіри голови, порівняно з плацебо.
Псоріатичний артрит
Під час трьох рандомізованих, подвійно сліпих, плацебо‑контрольованих досліджень ІІІ фази за участю пацієнтів з активним псоріатичним артритом (≥ 3 набряклих суглобів та ≥ 3 болючих суглобів), незважаючи на терапію нестероїдними протизапальними препаратами (НПЗП), кортикостероїдами або хворобомодифікуючими антиревматичними препаратами, безпеку та ефективність секукінумабу оцінювали у 1999 пацієнтів. У ці дослідження були включені пацієнти з кожним підтипом псоріатичного артриту, включаючи поліартикулярний артрит без ревматоїдних вузликів, спондиліт з периферичним артритом, асиметричний периферичний артрит, ураження дистальних міжфалангових суглобів та мутилюючий артрит. Під час досліджень пацієнти мали в анамнезі псоріатичний артрит протягом щонайменше п’яти років. У більшості пацієнтів також був діагностований активний псоріаз з ураженням шкіри або документально підтверджений псоріаз в анамнезі. Понад 61 % та 42 % пацієнтів із псоріатичним артритом на початку дослідження мали ентезит та дактиліт відповідно. Для всіх досліджень первинною кінцевою точкою була відповідь за критеріями ACR (Американської колегії ревматології) 20. Щодо дослідження псоріатичного артриту 1 та дослідження псоріатичного артриту 2 первинна кінцева точка була досягнута на 24-ому тижні. Щодо дослідження псоріатичного артриту 3 первинна кінцева точка була досягнута на 16-ому тижні разом з основною вторинною кінцевою точкою, зміною відносно вихідних показників за модифікованою шкалою Шарпа (mTSS) на 24-ому тижні.
В дослідженні псоріатичного артриту 1, дослідженні псоріатичного артриту 2 та дослідженні псоріатичного артриту 3 29 %, 35 % та 30 % пацієнтів раніше отримували терапію анти‑ФНПα засобом та припинили прийом анти-ФНПα засобу через відсутність ефективності або переносимості (пацієнти з недостатньою відповіддю на терапію анти-ФНПα).
В дослідженні псоріатичного артриту 1 (FUTURE 1) оцінювали 606 пацієнтів, з яких 60,7 % одночасно отримували метотрексат. Пацієнти, рандомізовані до групи секукінумабу, отримували 10 мг/кг внутрішньовенно на 0, 2 та 4-ому тижні, потім 75 мг або 150 мг підшкірно щомісяця, починаючи з 8 тижня. Пацієнти, рандомізовані до групи плацебо, які не відповіли на лікування на 16-ому тижні (препарат невідкладної допомоги), та інші пацієнти в групі плацебо на 24-ому тижні були переведені на секукінумаб (в дозі 75 мг або 150 мг підшкірно) з наступним щомісячним введенням аналогічної дози.
В дослідженні псоріатичного артриту 2 (FUTURE 2) оцінювали 397 пацієнтів, з яких 46,6 % одночасно отримували метотрексат. Пацієнти, рандомізовані до групи секукінумабу, отримували 75 мг, 150 мг або 300 мг підшкірно на 0, 1, 2, 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4 тижня. Пацієнти, рандомізовані до групи плацебо, які не відповіли на лікування на 16-ому тижні (рання відповідь), були переведені на секукінумаб (в дозі 150 мг або 300 мг підшкірно) на 16-ому тижні з наступним щомісячним введенням аналогічної дози. Пацієнти, рандомізовані до групи плацебо, які відповіли на лікування на 16-ому тижні, були переведені на секукінумаб (в дозі 150 мг або 300 мг підшкірно) на 24-ому тижні з наступним щомісячним введенням аналогічної дози.
В дослідженні псоріатичного артриту 3 (FUTURE 5) оцінювали 996 пацієнтів, з яких 50,1 % одночасно отримували метотрексат. Пацієнти були рандомізовані в групу секукінумабу в дозі 150 мг, 300 мг або плацебо підшкірно на 0, 1, 2, 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4 тижня, або щомісячної ін’єкції секукінумабу в дозі 150 мг (без навантаження). Пацієнти, рандомізовані до групи плацебо, які не відповіли на лікування на 16-ому тижні (рання відповідь), були переведені на секукінумаб (в дозі 150 мг або 300 мг підшкірно) на 16-ому тижні з наступним щомісячним введенням аналогічної дози. Пацієнти, рандомізовані до групи плацебо, які відповіли на лікування на 16-ому тижні, були переведені на секукінумаб (в дозі 150 мг або 300 мг підшкірно) на 24-ому тижні з наступним щомісячним введенням аналогічної дози.
Ознаки та симптоми
Лікування секукінумабом призвело до значного покращення показників активності захворювання у порівнянні з плацебо на 16 та 24-ому тижні (див. таблицю 4).
Таблиця 4 Клінічна відповідь у дослідженні псоріатичного артриту 2 та дослідженні псоріатичного артриту 3 на 16-мі та 24-му тижні
Показники | Дослідження псоріатичного артриту 2 | Дослідження псоріатичного артриту 3 | ||||
Плацебо | 150 мг1 | 300 мг1 | Плацебо | 150 мг1 | 300 мг1 | |
Кількість рандомізованих пацієнтів | 98 | 100 | 100 | 332 | 220 | 222 |
Відповідь ACR 20, n (%) | ||||||
Тиждень 16 | 18 (18,4 %) | 60 (60,0%***) | 57 (57,0%***) | 91◊ (27,4 %) | 122◊ (55,5%***) | 139◊ (62,6 %***) |
Тиждень 24 | 15◊ (15,3 %) | 51◊ (51,0%***) | 54◊ (54,0%***) | 78 (23,5 %) | 117 (53,2%***) | 141 (63,5 %***) |
Відповідь ACR 50, n (%) | ||||||
Тиждень 16 | 6 (6,1 %) | 37 (37,0%***) | 35 (35,0%***) | 27 (8,1 %) | 79 (35,9 %*) | 88 (39,6 %*) |
Тиждень 24 | 7 (7,1 %) | 35 (35,0 %) | 35 (35,0 %**) | 29 (8,7 %) | 86 (39,1%***) | 97 (43,7 %***) |
Відповідь ACR 70, n (%) | ||||||
Тиждень 16 | 2 (2,0 %) | 17 (17,0 %**) | 15 (15,0 %**) | 14 (4,2 %) | 40 (18,2%***) | 45 (20,3 %***) |
Тиждень 24 | 1 (1,0 %) | 21 (21,0 %**) | 20 (20,0 %**) | 13 (3,9 %) | 53 (24,1%***) | 57 (25,7 %***) |
DAS28‑CRP | ||||||
Тиждень 16 | -0,50 | -1,45*** | -1,51*** | -0,63 | -1,29* | -1,49* |
Тиждень 24 | -0,96 | -1,58** | -1,61** | -0,84 | -1,57*** | -1,68*** |
Кількість пацієнтів з ≥ 3 % псоріазом шкіри від ППТ на вихідному рівні | 43 (43,9 %) | 58 (58,0 %) | 41 (41,0 %) | 162 (48,8 %) | 125 (56,8 %) | 110 (49,5 %) |
Відповідь PASI 75, n (%) | ||||||
Тиждень 16 | 3 (7,0 %) | 33 (56,9%***) | 27 (65,9%***) | 20 (12,3 %) | 75 (60,0 %*) | 77 (70,0 %*) |
Тиждень 24 | 7 (16,3 %) | 28 (48,3 %**) | 26 (63,4%***) | 29 (17,9 %) | 80 (64,0%***) | 78 (70,9 %***) |
Відповідь PASI 90, n (%) | ||||||
Тиждень 16 | 3 (7,0 %) | 22 (37,9%***) | 18 (43,9%***) | 15 (9,3 %) | 46 (36,8 %*) | 59 (53,6 %*) |
Тиждень 24 | 4 (9,3 %) | 19 (32,8 %**) | 20 (48,8%***) | 19 (11,7 %) | 51 (40,8%***) | 60 (54,5 %***) |
Розвиток дактилітів, n (%) † | ||||||
Тиждень 16 | 10 (37 %) | 21 (65,6 %*) | 26 (56,5 %) | 40 (32,3 %) | 46 (57,5 %*) | 54 (65,9 %*) |
Тиждень 24 | 4 (14,8 %) | 16 (50,0 %**) | 26 (56,5 %**) | 42 (33,9 %) | 51 (63,8%***) | 52 (63,4 %***) |
Усунення ентезитів, n (%) ‡ | ||||||
Тиждень 16 | 17 (26,2 %) | 32 (50,0 %**) | 32 (57,1%***) | 68 (35,4 %) | 77 (54,6 %*) | 78 (55,7 %*) |
Тиждень 24 | 14 (21,5 %) | 27 (42,2 %*) | 27 (48,2 %**) | 66 (34,4 %) | 77 (54,6%***) | 86 (61,4 %***) |
*p < 0,05, **p < 0,01, ***p < 0,001 проти плацебо.
Всі p-значення коригуються з урахуванням кратності випробувань на підставі попередньо визначеної ієрархії на 24-ому тижні щодо дослідження псоріатичного артриту 2, за винятком ACR 70, дактиліту та ентезиту, які були пошуковими кінцевими точками та всіма кінцевими точками на 16-ому тижні.
Всі p-значення коригуються з урахуванням кратності випробувань на підставі попередньо визначеної ієрархії на 16-ому тижні щодо дослідження псоріатичного артриту 3, за винятком ACR 70, який був пошуковою кінцевою точкою та всіма кінцевими точками на 24-ому тижні.
Для відсутньої бінарної кінцевої точки використовується метод підстановки даних при відсутності відповіді.
ACR - Американська колегія ревматології; PASI - індекс площі та тяжкості псоріазу; DAS - індекс активності захворювання; ППТ - площа поверхні тіла.
◊Первинна кінцева точка.
1 Секукінумаб 150 мг або 300 мг підшкірно на 0, 1, 2, 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4 тижня.
†У пацієнтів з дактилітом на початку дослідження (n=27, 32, 46, відповідно щодо дослідження псоріатичного артриту 2 та n=124, 80, 82, відповідно щодо дослідження псоріатичного артриту 3).
‡У пацієнтів з ентезитом на початку дослідження (n=65, 64, 56, відповідно щодо дослідження псоріатичного артриту 2 та n=192, 141, 140, відповідно щодо дослідження псоріатичного артриту 3).
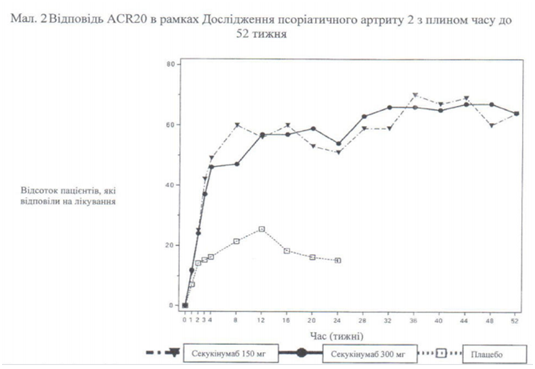
Початок дії секукінумабу спостерігається вже на 2 тижні. Статистично значуща різниця ACR 20 між групою секукінумабу і плацебо досягається на 3 тижні.
Кількість пацієнтів, які досягли відповіді ACR 20 відповідно до візиту, показана на рис. 2.
Аналогічні відповіді щодо первинних та основних вторинних кінцевих точок спостерігались у пацієнтів із псоріатичним артритом незалежно від одночасного застосування метотрексату. В дослідженні псоріатичного артриту 2 на 24-му тижні у пацієнтів в групі комбінованого застосування секукінумабу і метотрексату зареєстрована більш висока відповідь ACR 20 (47,7 % та 54,4 % пацієнтів в групі прийому дози 150 мг та 300 мг порівняно з 20,0 % в групі плацебо) та відповідь ACR 50 (31,8 % та 38,6 % пацієнтів в групі прийому дози 150 мг та 300 мг порівняно з 8,0 % в групі плацебо). У пацієнтів в групі секукінумабу без одночасного застосування метотрексату зареєстрована більш висока відповідь ACR 20 (53,6 % та 53,6 % пацієнтів у групі прийому дози 150 мг та 300 мг порівняно з 10,4 % в групі плацебо) та відповідь ACR 50 (37,5 % та 32,1 % пацієнтів у групі прийому дози 150 мг та 300 мг порівняно з 6,3 % в групі плацебо).
В дослідженні псоріатичного артриту 2 як у пацієнтів, які не отримували терапію анти‑ФНПα, так і у пацієнтів з недостатньою відповіддю на терапію анти‑ФНПα, які приймали секукінумаб, відзначалась більш висока відповідь ACR 20 порівняно з плацебо на 24-му тижні, при цьому дещо вища відповідь спостерігалась в групі пацієнтів, які не отримували терапію анти‑ФНПα (пацієнти, які не отримували терапію анти‑ФНПα: 64 % та 58 % - в групі прийому дози 150 мг та 300 мг порівняно з 15,9 % в групі плацебо; пацієнти з недостатньою відповіддю на терапію анти‑ФНПα: 30 % та 46 % в групі прийому дози 150 мг та 300 мг порівняно з 14,3 % в групі плацебо). В підгрупі пацієнтів з недостатньою відповіддю на терапію анти‑ФНПα тільки прийом дози 300 мг продемонстрував значно вищу частоту відповіді ACR 20 порівняно з плацебо (p < 0,05), а також клінічно значущі переваги, ніж прийом дози 150 мг, щодо декількох вторинних кінцевих точок. Покращення за індексом PASI 75 спостерігались в обох підгрупах, а прийом дози 300 мг показав статистично значущі переваги у пацієнтів з недостатньою відповіддю на терапію анти‑ФНПα.
Кількість пацієнтів із псоріатичним артритом з ураженням осьового скелета була дуже незначною, що не дає змоги дати обґрунтовану оцінку.
Покращення спостерігались за усіма показниками шкали ACR, включаючи оцінку болю пацієнтами. Під час дослідження псоріатичного артриту 2 кількість пацієнтів, які досягли відповіді за модифікованими критеріями відповіді псоріатичного артриту (PsARC), була вищою в групі секукінумабу (59,0 % та 61,0 % в групі прийому дози 150 мг та 300 мг), ніж в групі плацебо (26,5 %), на 24-му тижні.
В дослідженні псоріатичного артриту 1 та дослідженні псоріатичного артриту 2 ефективність зберігалась до 104-го тижня. В дослідженні псоріатичного артриту 2 серед 200 пацієнтів, які спочатку були рандомізовані до групи секукінумабу в дозі 150 мг та 300 мг, 178 (89 %) все ще отримували лікування на 52-му тижні. Зі 100 пацієнтів, рандомізованих до групи секукінумабу в дозі 150 мг, 64, 39 та 20 осіб досягли відповіді ACR 20/50/70 відповідно. Зі 100 пацієнтів, рандомізованих до групи секукінумабу в дозі 300 мг, 64, 44 та 24 особи досягли відповіді ACR 20/50/70 відповідно.
Рентгенологічна відповідь
У дослідженні псоріатичного артриту 3 пригнічення прогресування структурного ураження оцінювалось рентгенографічно та визначалось за модифікованою шкалою Шарпа (mTSS) та її компонентами, показником ерозій та звуження суглобової щілини. Рентгенологічні знімки рук, зап’ястків та ніг були зроблені на початку дослідження, на 16-му тижні та/або 24-му тижні та оцінювались незалежно щонайменше двома фахівцями, яким не було надано інформації про групи лікування та номер візиту. Секукінумаб в дозі 150 мг та 300 мг значно пригнічував швидкість прогресування ураження периферичного суглоба порівняно з плацебо, розраховану як зміна відносно вихідних показників за mTSS на 24-му тижні (таблиця 5).
Пригнічення прогресування структурного ураження також оцінювалось під час дослідження псоріатичного артриту 1 на 24-му та 52-му тижні відносно вихідних показників. Дані на 24-тижні представлено в таблиці 5.
Таблиця 5 Зміни показника модифікованої шкали Шарпа при псоріатичному артриті
Показники | Дослідження псоріатичного артриту 3 | Дослідження псоріатичного артриту 1 | |||
плацебо n = 296 | секукінумаб 150 мг1 n = 213 | секукінумаб 300 мг1 n = 217 | плацебо n = 179 | секукінумаб 150 мг2 N = 185 | |
Загальна кількість балів | |||||
Вихідний рівень (SD) | 15,0 (38,2) | 13,5 (25,6) | 12,9 (23,8) | 28,4 (63,5) | 22,3 (48,0) |
Середня зміна на 24-ому тижні | 0,50 | 0,13* | 0,02* | 0,57 | 0,13* |
*p < 0,05 на підставі номінального, але не скоригованого p-значення.
1Секукінумаб в дозі 150 мг або 300 мг підшкірно на 0, 1, 2, 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4 тижня.
210 мг/кг на 0, 2 тижні з наступним введенням підшкірної дози 75 мг або 150 мг, починаючи з 4 тижня.
В дослідженні псоріатичного артриту 1 пригнічення структурного ураження зберігалось до 52-го тижня при прийомі секукінумабу.
В дослідженні псоріатичного артриту 3 кількість пацієнтів з відсутністю прогресування захворювання (визначено, як зміна відносно вихідних показників за шкалою mTSS ≤ 0,5) від початку рандомізації до 24-го тижня становила 80,3 %, 88,5 % та 73,6 % в групі прийому секукінумабу в дозі 150 мг, 300 мг та плацебо. Вплив пригнічення структурного ураження спостерігався у пацієнтів, які не отримували терапію анти‑ФНПα, та пацієнтів з недостатньою відповіддю на терапію анти‑ФНПα, а також у пацієнтів, які одночасно приймали або не приймали метотрексат.
В дослідженні псоріатичного артриту 1 кількість пацієнтів з відсутністю прогресування захворювання (визначено, як зміна відносно вихідних показників за шкалою mTSS ≤ 0,5) від початку рандомізації до 24-го тижня становила 82,3 % в групі прийому секукінумабу в дозі 10 мг/кг внутрішньовенно - 150 мг підшкірно як підтримуючої дози та 75,7 % в групі плацебо. Кількість пацієнтів з відсутністю прогресування з 24-го тижня по 52-й тиждень в групі внутрішньовенного введення секукінумабу в дозі 10 мг/кг з наступним введенням дози 150 мг підшкірно як підтримуючої дози та в групі плацебо, які були переведені на дозу 75 мг або 150 мг підшкірно кожні 4 тижні на 16-ому тижні або 24-ому тижні, становила 85,7 % та 86,8 %.
Якість життя, пов’язана з функціональним статусом і станом здоров’я
Під час дослідження псоріатичного артриту 2 та дослідження псоріатичного артриту 3 пацієнти, які отримували секукінумаб в дозі 150 мг (p = 0,0555 та p < 0,0001) та 300 мг (p = 0,0040 та p < 0,0001), продемонстрували покращення функціонального статусу порівняно з пацієнтами, які отримували плацебо, відповідно до індексу порушення життєдіяльності, підрахованого за опитувальником для оцінки стану здоров’я (HAQ‑DI), на 24-му та 16-му тижнях. Покращення показників відповідно до HAQ‑DI спостерігалось, незважаючи на попередню терапію анти‑ФНПα. Аналогічні відповіді відмічені в дослідженні псоріатичного артриту 1.
Пацієнти, які приймали секукінумаб, відмічали значуще покращення пов’язаної зі станом здоров’я якості життя, розраховане за скороченою формою шкали неспецифічного опитувальника для оцінки фізичного компонента здоров’я (SF‑36 PCS) (p < 0,001). Також спостерігалось статистично значуще покращення щодо пошукових кінцевих точок за шкалою функціональної оцінки терапії хронічного захворювання-втоми (FACIT-F) при прийомі 150 мг та 300 мг порівняно з плацебо (7,97, 5,97 проти 1,63). В дослідженні псоріатичного артриту 2 ці покращення зберігались до 104-го тижня.
Подібні відповіді були відмічені в дослідженні псоріатичного артриту 1, а ефективність зберігалася до 52-го тижня.
Аксіальний спондилоартрит
Анкілозуючий спондиліт (AC)/ рентгенологічний аксіальний спондилоартрит
Під час двох рандомізованих, подвійно сліпих, плацебо‑контрольованих досліджень III фази за участю пацієнтів з активним анкілозуючим спондилітом з показниками відповідно до Батського індексу активності анкілозуючого спондиліту (BASDAI) ≥ 4, незважаючи на терапію нестероїдними протизапальними препаратами (НПЗП), кортикостероїдами або хворобомодифікуючими антиревматичними препаратами, безпеку та ефективність секукінумабу оцінювали у 816 пацієнтів. В ході дослідження анкілозуючого спондиліту 1 та дослідження анкілозуючого спондиліту 2 пацієнтам був діагностований анкілозуючий спондиліт з медіаною тривалості 2,7-5,8 року. В обох цих дослідженнях первинною кінцевою точкою було щонайменше 20 % покращення за критеріями міжнародного товариства з вивчення спондилоартритів (ASAS 20) на 16-ому тижні.
В дослідженні анкілозуючого спондиліту 1, дослідженні анкілозуючого спондиліту 2 та дослідженні анкілозуючого спондиліту 3 27,0 %, 38,8 % та 23,5 % пацієнтів раніше отримували терапію анти‑ФНПα засобом та припинили прийом анти-ФНПα засобу через відсутність ефективності або непереносимість (пацієнти з недостатньою відповіддю на терапію анти-ФНПα).
В дослідженні анкілозуючого спондиліту 1 (MEASURE 1) оцінювали 371 пацієнта, з яких 14,8 % та 33,4 % одночасно застосовували метотрексат або сульфасалазин. Пацієнти, рандомізовані до групи секукінумабу, отримували 10 мг/кг внутрішньовенно на 0, 2 та 4 тижні з наступним щомісячним введенням дози 75 мг або 150 мг підшкірно, починаючи з
8-го тижня. Пацієнти, рандомізовані до групи плацебо, не відповіли на лікування на 16-ому тижні (рання відповідь), всі інші пацієнти в групі плацебо на 24-му тижні були переведені на секукінумаб (в дозі 75 мг або 150 мг підшкірно) з наступним щомісячним введенням аналогічної дози.
В дослідженні анкілозуючого спондиліту 2 (MEASURE 2) оцінювали 219 пацієнтів, з яких 11,9 % та 14,2 % одночасно застосовували метотрексат або сульфасалазин. Пацієнти, рандомізовані до групи секукінумабу, отримували 75 мг або 150 мг підшкірно на 0, 1, 2, 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4 тижня. На 16-ому тижні пацієнти, які були рандомізовані до групи плацебо на початку дослідження, були повторно рандомізовані до групи секукінумабу (в дозі 75 мг або 150 мг підшкірно) при щомісячному режимі введення.
В дослідженні анкілозуючого спондиліту 3 (MEASURE 3) оцінювали 226 пацієнтів, з яких 13,3 % та 23,5 % одночасно застосовували метотрексат або сульфасалазин. Пацієнти, рандомізовані до групи секукінумабу, отримували 10 мг/кг внутрішньовенно на 0, 2 та 4 тижні з наступним введенням дози 150 мг або 300 мг підшкірно щомісяця. На 16-ому тижні пацієнти, які були рандомізовані до групи плацебо на початку дослідження, були повторно рандомізовані до групи секукінумабу (в дозі 150 мг або 300 мг підшкірно) зі щомісячним режимом введення. Первинною кінцевою точкою була відповідь ASAS 20 на 16-му тижні. Пацієнти отримували лікування у сліпому режимі до 52-го тижня. Дослідження продовжувалось до 156 тижня.
Ознаки та симптоми
У дослідженні анкілозуючого спондиліту 2 лікування секукінумабом в дозі 150 мг призводило до значного зменшення активності захворювання порівняно з плацебо на 16-ому тижні (див. таблицю 6).
Таблиця 6
Клінічна відповідь в рамках дослідження анкілозуючого спондиліту 2 на 16-ому тижні
Результат (p-значення проти плацебо) | Плацебо (n = 74) | Секукінумаб | |
75 мг (n = 73) | 150 мг (n = 72) | ||
Відповідь ASAS 20, % | 28,4 | 41,1 | 61,1*** |
Відповідь ASAS 40, % | 10,8 | 26,0 | 36,1*** |
hsCRP, (пост‑BSL/BSL співвідношення) | 1,13 | 0,61 | 0,55*** |
ASAS 5/6, % | 8,1 | 34,2 | 43,1*** |
Часткова ремісія ASAS, % | 4,1 | 15,1 | 13,9 |
BASDAI 50, % | 10,8 | 24,7* | 30,6** |
Значне покращення ASDAS-CRP | 4,1 | 15,1* | 25,0*** |
*p < 0,05, **p < 0,01, ***p < 0,001 проти плацебо.
Всі p-значення коригуються з урахуванням кратності випробувань на підставі попередньо визначеної ієрархії, окрім BASDAI 50 та ASDAS-CRP.
Для відсутньої бінарної кінцевої точки використовується метод підстановки даних при відсутності відповіді.
ASAS - критерії міжнародного товариства з вивчення спондилоартритів; BASDAI - Батський індекс активності анкілозуючого спондиліту; hsCRP - високочутливий С-реактивний протеїн; ASDAS - шкала активності анкілозуючого спондиліту; BSL - вихідний рівень.
Під час дослідження анкілозуючого спондиліту 2 початок дії секукінумабу в дозі 150 мг спостерігався вже на 1-му тижні щодо ASAS 20 та на 2-му тижні щодо ASAS 40 (більш ефективний, ніж плацебо).
При прийомі секукінумабу в дозі 150 мг порівняно з плацебо відповідь ASAS 20 була вищою: покращилась на 16-ому тижні і у пацієнтів, які не отримували терапію анти‑ФНПα (68,2 % проти 31,1 %; p < 0,05), і у пацієнтів з недостатньою відповіддю на терапію анти‑ФНПα (50,0 % проти 24,1 %; p < 0,05).
Під час кожного дослідження анкілозуючого спондиліту пацієнти, які приймали секукінумаб (150 мг в рамках дослідження анкілозуючого спондиліту 2 та обидва режими в рамках дослідження анкілозуючого спондиліту 1), продемонстрували значне зменшення ознак та симптомів на 16-ому тижні з порівнянною величиною відповіді та ефективністю, що зберігались до 52-го тижня, у пацієнтів, які не отримували терапію анти‑ФНПα, та пацієнтів з недостатньою відповіддю на терапію анти‑ФНПα. У дослідженні анкілозуючого спондиліту 2 із 72 пацієнтів, які спочатку були рандомізовані до групи секукінумабу в дозі 150 мг, 61 (84,7 %) все ще отримували лікування на 52-му тижні. З 72 пацієнтів, рандомізованих до групи секукінумабу в дозі 150 мг, у 45 та 35 пацієнтів зафіксована відповідь АСАС 20 та 40 відповідно.
Під час дослідження анкілозуючого спондиліту 3 у пацієнтів, які отримували секукінумаб (в дозі 150 мг та 300 мг), спостерігалось покращення ознак та симптомів і збереження ефективності, незважаючи на дозу, порівняно із плацебо на 16-му тижні щодо первинної кінцевої точки (ASAS 20). Загалом, частота збереження ефективності в групі прийому 300 мг була значно вищою, ніж в групі прийому 150 мг, щодо вторинних кінцевих точок. Під час сліпого періоду відповіді за критеріями ASAS 20 та ASAS 40 становили 69,7 % та 47,6 % відповідно у групі прийому 150 мг та 74,3 % і 57,4 % відповідно в групі прийому 300 мг на 52-му тижні. Відповіді ASAS 20 та ASAS 40 зберігались до 156-го тижня (69,5 % та 47,6 % відповідно в групі прийому 150 мг проти 74,8 % та 55,6 % відповідно в групі прийому 300 мг). Більша частота відповіді, що спостерігалась в групі застосування 300 мг, також відзначалась стосовно часткової ремісії (ASAS PR) на 16-му тижні та підтримувалась до 156-го тижня. Відмінності щодо більшої частоти відповідей, що спостерігались в групі застосування 300 мг, порівняно з групою 150 мг, відзначались у пацієнтів, які отримували анти-ФНПα терапію (n = 36), порівняно з пацієнтами, які не отримували анти-ФНПα терапію (n = 114).
Рухливість хребта
Пацієнти, які отримували секукінумаб в дозі 150 мг, продемонстрували покращення рухливості хребта, оціненої за зміною порівняно з вихідним рівнем індексу порушень рухів в хребті при АС (BASMI) на 16-ому тижні в рамках дослідження анкілозуючого спондиліту 1 (‑0,40 проти ‑0,12 в групі плацебо; p = 0,0114) та дослідження анкілозуючого спондиліту 2 (‑0,51 проти ‑0,22 в групі плацебо; p = 0,0533). Такі покращення зберігались до 52-го тижня.
Якість життя, пов’язана з функціональним статусом і станом здоров’я
Під час дослідження анкілозуючого спондиліту 1 та дослідження анкілозуючого спондиліту 2 пацієнти, які отримували секукінумаб в дозі 150 мг, продемонстрували покращення пов’язаної зі станом здоров’я якості життя, розраховане за шкалою опитувальника для оцінки якості життя при анкілозуючому спондиліті (ASQoL) (p=0,001) та шкалою SF-36 фізичного компонента здоров’я (SF-36PCS) (p<0.001). Пацієнти, які отримували секукінумаб в дозі 150 мг, також продемонстрували статистично значуще покращення щодо пошукових кінцевих точок функціонального статусу відповідно до Батського індексу функціональних порушень при анкілозуючому спондиліті (BASFI) порівняно з плацебо (‑2,15 проти ‑0,68), а також втоми за шкалою функціональної оцінки терапії хронічного захворювання-втоми (FACIT-втома) порівняно з плацебо (8,10 проти 3,30). Такі покращення зберігались до 52-го тижня.
Нерентгенологічний аксіальний спондилоартрит
Безпеку та ефективність секукінумабу оцінювали під час одного рандомізованого, подвійно сліпого, плацебо-контрольованого дослідження фази ІІІ (PREVENT), що складається з дворічної основної фази та дворічної розширеної фази, за участю 555 пацієнтів з активним нерентгенологічним аксіальним спондилоартритом, які відповідали критеріям класифікації Міжнародного товариства з дослідження спондилоартриту (ASAS), за відсутності рентгенологічних доказів зміни крижово-клубового суглоба, що відповідало Модифікованим нью-йоркським критеріям для діагностики анкілозуючого спондиліту (АС). У пацієнтів, які брали участь у дослідженні, встановлювалось захворювання в активній фазі зі значенням Індексу активності захворювання анкілозуючим спондилітом (BASDAI), що дорівнює ≥ 4, за візуальною аналоговою шкалою болю в спині на рівні ≥ 40 (в діапазоні 0 або 100 мм), незважаючи на поточну або попередню терапію нестероїдними протизапальними препаратами (НПЗП) та підвищений рівень високочутливого С-реактивного білка та/або докази розвитку сакроілітиту на МРТ. Пацієнтам в рамках цього дослідження діагностували аксіальний спондилоартрит протягом в середньому 2,1-3,0 року. 54 % учасників дослідження були жінками.
Під час дослідження PREVENT 9,7 % пацієнтів раніше отримували терапію анти-ФНПα засобом та припинили прийом анти-ФНПα засобу через відсутність ефективності або непереносимість (пацієнти з недостатньою відповіддю на терапію анти-ФНПα).
Під час дослідження PREVENT 9,9 % та 14,8 % пацієнтів застосовували одночасно метотрексат або сульфасалазин відповідно. Протягом подвійно сліпого періоду дослідження пацієнти отримували плацебо або секукінумаб протягом 52 тижнів. Пацієнти були рандомізовані в групу підшкірного введення секукінумабу в дозі 150 мг на 0, 1, 2, 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4 тижня, або в групу разового щомісячного введення секукінумабу в дозі 150 мг. Первинною кінцевою точкою було щонайменше 40 % покращення за критеріями Міжнародного товариства з дослідження спондилоартриту (ASAS 40) на 16-му тижні у пацієнтів, які не отримували терапію анти- ФНПα.
Ознаки та симптоми
Під час дослідження PREVENT лікування секукінумабом в дозі 150 мг призводило до значущого зниження показників активності захворювання порівняно з плацебо на 16-му тижні. Ці виміри включали оцінки відповідей за критеріями ASAS 40, ASAS 5/6, BASDAI, BASDAI 50, оцінку високочутливого С-реактивного білка (hsCRP), ASAS 20, а також оцінку часткової ремісії ASAS порівняно з такими при застосуванні плацебо. Відповіді зберігались до 52-го тижня.
Під час дослідження PREVENT початок дії секукінумабу в дозі 150 мг спостерігався вже на 3-му тижні щодо ASAS 40 у пацієнтів, які не отримували терапію анти-ФНПα (більш ефективний, ніж плацебо).
У пацієнтів з недостатньою відповіддю на терапію анти-ФНПα в групі секукінумабу в дозі 150 мг відповіді ASAS 40 також покращувались на 16-му тижні порівняно з групою плацебо.
Фізична функція та якість життя, пов’язана зі станом здоров’я
Пацієнти, які приймали секукінумаб в дозі 150 мг, відмічали статистично значущі покращення фізичної функції до 16-го тижня, розраховані за допомогою функціонального індексу активності анкілозуючого спондиліту (BASFI) (16-й тиждень: 1,75 проти 1,01, p < 0,05), порівняно з пацієнтами, які отримували плацебо. Пацієнти, які приймали секукінумаб, відмічали значущі покращення якості життя, пов’язаної зі станом здоров’я, до 16-го тижня, розраховані за допомогою опитувальника ASQoL (середня зміна, визначена методом найменших квадратів: 16-й тиждень: 3,45 проти 1,84, p < 0,05) та за шкалою неспецифічного опитувальника для оцінки фізичного компонента здоров’я (SF-36 PCS) (середня зміна, визначена методом найменших квадратів: 16-й тиждень: 5,71 проти 2,93, p < 0,05), порівняно з пацієнтами, які отримували плацебо. Ці покращення зберігались до 52-го тижня.
Рухливість хребта
Рухливість хребта оцінювали відповідно до індексу порушень рухів в хребті при АС (BASMI) на 16-ому тижні. Чисельно вищі покращення були продемонстровані у пацієнтів, які отримували секукінумаб, порівняно з пацієнтами, які отримували плацебо, на 4, 8, 12 та 16 тижнях.
Пригнічення запалення за даними магнітно-резонансної томографії (МРТ)
Ознаки запалення оцінювали за допомогою МРТ на початковому рівні та на 16-му тижні; вони були виражені як зміна на початковому рівні за Берлінською шкалою оцінки набряків крижово-клубового суглоба, шкалою ASspiMRI-a та Берлінською шкалою оцінки хребта. У пацієнтів, які отримували секукінумаб, спостерігалось пригнічення ознак запалення як в крижово-клубових суглобах, так і в хребті. Середня зміна відносно вихідного рівня за Берлінською шкалою оцінки набряків крижово-клубового суглоба становила 1,68 у пацієнтів, які отримували секукінумаб в дозі 150 мг (n = 180), проти 0,39 у пацієнтів, які отримували плацебо (n = 174) (p < 0,05).
Діти
Бляшковий псоріаз у дітей
Встановлено, що секукінумаб послаблює ознаки та симптоми захворювання, а також покращує якість життя, пов’язану зі здоров’ям, у дітей віком від 6 років із бляшковим псоріазом
Тяжкий бляшковий псоріаз
Безпеку та ефективність секукінумабу оцінювали під час рандомізованого, подвійно сліпого, плацебо- та етанерцепт-контрольованого дослідження фази III за участю педіатричних пацієнтів віком від 6 до < 18 років із тяжким бляшковим псоріазом, як визначено за шкалою PASI ≥ 20, за IGA mod 2011 більше 4 балів і BSA ≥ 10 %, які були кандидатами на системну терапію. Приблизно 43 % пацієнтів раніше отримували фототерапію, 53 % - звичайну системну терапію, 3 % - імунобіологічні препарати, 9 % - супутню терапію псоріатичного артриту.
Під час дослідження 1 псоріазу у дітей оцінювали 162 пацієнтів, які були рандомізовані для отримання низьких доз секукінумабу (75 мг при масі тіла < 50 кг або 150 мг при масі тіла ≥ 50 кг), високих доз секукінумабу (75 мг при масі тіла < 25 кг, 150 мг при масі тіла від ≥ 25 кг до < 50 кг або 300 мг при масі тіла ≥ 50 кг) або плацебо на 0, 1, 2, 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4 тижня, чи етанерцепту. Пацієнти, рандомізовані для приймання етанерцепту, отримували 0,8 мг/кг на тиждень (максимум до 50 мг).
Пацієнти, рандомізовані для приймання плацебо, якіне відповідали на лікування, на 12-му тижні були переведені в групу прийому низьких або високих доз секукінумабу (дозування для групи пацієнтів, розподілених за масою тіла) та отримували досліджуваний лікарський засіб на 12, 13, 14 та 15 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 16 тижня. Первинними кінцевими точками були кількість пацієнтів, які досягли індексу PASI 75 та IGA mod 2011 «чисто» або «майже чисто» (0 або 1) на 12-му тижні.
Протягом 12-тижневого плацебо-контрольованого періоду ефективність низьких та високих доз секукінумабу була порівнянною у первинних кінцевих точках. Оцінки відношення шансів на користь обох доз секукінумабу були статистично значущими щодо індексів PASI 75 та IGA mod 2011 0 або 1.
За ефективністю та безпекою усіх пацієнтів спостерігали протягом 52 тижнів після прийому першої дози. Кількість пацієнтів, які досягли індексу PASI 75 та IGA mod 2011 «чисто» або «майже чисто» (0 або 1), свідчила про розділення між групами лікування секукінумабом та плацебо під час першого візиту на початку дослідження на 4-му тижні, при цьому різниця стає більш вираженою на 12-му тижні. Відповідь зберігалася протягом 52 тижнів. Поліпшення частоти відповіді PASI 50, 90, 100 та показників дерматологічного індексу якості життя дітей (CDLQI) 0 або 1 також зберігалися протягом 52 тижнів.
Крім цього, частота відповіді PASI 75, IGA 0 або 1, PASI 90 на 12-му та 52-му тижнях в обох групах прийому низьких та високих доз секукінумабу була вищою, ніж частота, що спостерігалась у пацієнтів, які отримували етанерцепт.
По закінченні 12 тижня ефективність як низьких, так і високих доз секукінумабу була порівнянна, хоча ефективність високої дози була вищою у пацієнтів з масою тіла ≥ 50 кг. Профілі безпеки низьких та високих доз були порівнянними та відповідали профілю безпеки у дорослих.
У більшої кількості педіатричних пацієнтів, які отримували секукінумаб, спостерігалось покращення якості життя, пов’язаної зі здоров’ям, виміряної за шкалою CDLQI (0 або 1), порівняно з плацебо на 12-му тижні (група прийому низьких доз - 44,7 %, група прийому високих доз - 50 %, група прийому плацебо - 15 %). З плином часу, включаючи 52 тиждень, частка пацієнтів з покращенням в обох групах секукінумабу була чисельно більша, ніж в групі етанерцепту (група прийому низьких доз - 60,6 %, група прийому високих доз - 66,7 %, група прийому етанерцепту - 44,4 %).
Помірний або тяжкий бляшковий псоріаз
За прогнозами, секукінумаб є ефективним для лікування дітей з помірним бляшковим псоріазом на підставі продемонстрованої ефективності та причинно-наслідкового зв’язку у дорослих пацієнтів з помірним або тяжким бляшковим псоріазом, а також подібності перебігу захворювання, патофізіології та дії лікарського засобу у дорослих та дітей при однакових рівнях експозиції.
Більше того, ефективність та безпеку секукінумабу оцінювали під час відкритого, в двох паралельних групах багатоцентрового дослідження фази III за участю педіатричних пацієнтів віком від 6 до < 18 років із помірним або тяжким бляшковим псоріазом, визначеним за шкалою PASI ≥ 12, показником IGA mod 2011 ≥ 3 та BSA ≥ 10 %, які були кандидатами на системну терапію.
Під час дослідження 2 псоріазу у дітей досліджували 84 пацієнтів, які були рандомізовані для отримання низьких доз секукінумабу (75 мг при масі тіла < 50 кг або 150 мг при масі тіла ≥ 50 кг) або високих доз секукінумабу (75 мг при масі тіла < 25 кг, 150 мг при масі тіла від ≥ 25 кг до < 50 кг або 300 мг при масі тіла ≥ 50 кг) на 0, 1, 2, 3 тижні з наступним щомісячним введенням аналогічної дози, починаючи з 4 тижня.
Комбінованими первинними кінцевими точками були кількість пацієнтів, які досягли показників PASI 75 та IGA mod 2011 «чисто» або «майже чисто» (0 або 1) на 12-му тижні.
Ефективність низьких та високих доз секукінумабу була порівнянною та показала статистично значуще покращення порівняно з групою плацебо у комбінованих первинних кінцевих точках. Оцінена на основі досвіду ймовірність позитивного ефекту лікування становила 100 %.
Щодо ефективності за станом усіх пацієнтів спостерігали протягом щонайменше 24 тижнів після прийому першої дози. Ефективність (визначена як індекс PASI 75 та IGA mod 2011 «чисто» або «майже чисто» [0 або 1]) спостерігалась ще під час першого візиту після вихідного рівня на 2-му тижні, і кількість пацієнтів, які досягли відповіді PASI 75 та IGA mod 2011 «чисто» або «майже чисто» (0 або 1) зросла протягом 24-тижневого періоду. Покращення відповіді PASI 90 та PASI 100 також спостерігалось на 12-му тижні, що збільшувалось протягом 24-тижневого періоду.
Після 12-го тижня ефективність як низьких, так і високих доз секукінумабу була зіставна. Профілі безпеки низьких доз та високих доз були зіставними та відповідали профілю безпеки у дорослих.
Результати, отримані у педіатричних пацієнтів з помірним або тяжким бляшковим псоріазом, підтвердили прогнозовані припущення щодо ефективності та причинно-наслідкового зв’язку, які ґрунтуються на результатах, отриманих у дорослих пацієнтів.
У групі прийому низьких доз 50 % та 70,7 % пацієнтів досягли показників індексу CDLQI 0 або 1 через 12 та 24 тижні відповідно. У групі прийому високих доз 61,9 % та 60,5 % досягли показників індексу CDLQI 0 або 1 через 12 та 24 тижні відповідно.
Європейським агентством з лікарських засобів було відмовлено у представлені результатів досліджень секукінумабу в усіх пацієнтів дитячого віку від народження до 6 років з бляшковим псоріазом та пацієнтів дитячого віку від народження до 2 років з хронічним ідіопатичним артритом (див. розділ «Спосіб застосування та дози» для отримання інформації про застосування пацієнтам дитячого віку).
Європейським агентством з лікарських засобів було відмовлено у представлені результатів досліджень секукінумабу в усіх пацієнтів дитячого віку віком від 6 років до 18 років з бляшковим псоріазом та пацієнтів дитячого віку віком від 2 років до 18 років з хронічним ідіопатичним артритом (див. розділ «Спосіб застосування та дози» для отримання інформації про застосування пацієнтам у пацієнтів дитячого віку).
Фармакокінетика.
Більшість фармакокінетичних властивостей, що спостерігались у пацієнтів з бляшковим псоріазом, псоріатичним артритом та анкілозуючим спондилітом, були однаковими.
Абсорбція
Після одноразового підшкірного введення дози 300 мг у вигляді рідини здоровим добровольцям максимальна концентрація секукінумабу в сироватці крові становила 43,2±10,4 мкг/мл в інтервалі між 2 і 14 днем після введення.
На підставі популяційного фармакокінетичного аналізу після одноразового підшкірного введення дози 150 мг або 300 мг пацієнтам з бляшковим псоріазом максимальна концентрація секукінумабу в сироватці крові становила 13,7±4,8 мкг/мл або 27,3±9,5 мкг/мл в інтервалі між 5 та 6 днем після введення.
На підставі популяційного фармакокінетичного аналізу після початкового щотижневого введення під час першого місяця максимальна концентрація досягалася між 31 і 34 днем.
З огляду на дані моделювання, пікові концентрації в рівноважному стані (Cmax,ss) після підшкірного введення доз 150 мг і 300 мг становили відповідно 27,6 мкг/мл і 55,2 мкг/мл. Дані популяційного фармакокінетичного аналізу свідчать про те, що рівноважний стан досягається через 20 тижнів щомісячного введення.
У порівнянні з експозицією після одноразової дози у популяційному фармакокінетичному аналізі відзначено дворазове підвищення максимальної концентрації в сироватці крові і площі під фармакокінетичною кривою «концентрація-час» (AUC) після багаторазового щомісячного введення під час підтримуючої терапії.
Дані популяційного фармакокінетичного аналізу показали, що секукінумаб всмоктувався з середньою абсолютною біодоступністю 73 % у пацієнтів з бляшковим псоріазом. Серед досліджень, абсолютна біодоступність коливається в межах від 60 до 77 %, де вона розрахована.
На підставі популяційної фармакокінетичної моделі біодоступність секукінумабу у пацієнтів із псоріатичним артритом становила 85 %.
Розподіл
Середній об’єм розподілу в термінальній фазі (Vz) після одноразового внутрішньовенного введення варіював між 7,10 і 8,60 л у пацієнтів з бляшковим псоріазом, що дає змогу припустити, що секукінумаб обмежено розподіляється на периферії.
Біотрансформація
Більша частина елімінації IgG опосередковується внутрішньоклітинним катаболізмом з подальшою рідкою фазою або рецепторопосередкованим ендоцитозом.
Виведення
Середній системний кліренс (CL) після одноразового внутрішньовенного введення у пацієнтів із бляшковим псоріазом становить від 0,13 до 0,36 літрів/добу. У популяційному фармакокінетичному аналізі CL становив 0,19 л/добу у пацієнтів із бляшковим псоріазом. CL не залежав від статі пацієнта. Кліренс не залежав від дози та частоти застосування.
Середній період напіввиведення, розрахований в популяційному фармакокінетичному аналізі, становив 27 днів для пацієнтів із бляшковим псоріазом та коливався від 18 до 46 днів у різних псоріатичних дослідженнях із внутрішньовенним введенням.
Лінійність/нелінійність
Фармакокінетичні параметри при одноразовому і багаторазовому введенні секукінумабу у пацієнтів з бляшковим псоріазом були визначені в декількох дослідженнях з внутрішньовенним введенням в дозах від 1 × 0,3 мг/кг до 3 × 10 мг/кг і з підшкірним введенням в дозах від 25 мг одноразово до багаторазової дози 300 мг. При всіх режимах дозування експозиція була пропорційна дозі.
Особливі групи пацієнтів
Пацієнти літнього віку
Дані популяційного фармакокінетичного аналізу з обмеженою кількістю пацієнтів літнього віку (n = 71 для пацієнтів віком ≥ 65 років та n = 7 для пацієнтів віком ≥ 75 років) показали, що кліренс у пацієнтів літнього віку та пацієнтів віком до 65 років однаковий.
Пацієнти з порушенням функції нирок або печінки
Фармакокінетичні дані у пацієнтів з порушенням функції нирок або печінки відсутні. Нирковий кліренс незміненого секукінумабу, моноклонального антитіла IgG, є низьким та мінімальним. IgGs виводяться переважно шляхом катаболізму, і не очікується, що порушення функції печінки впливатиме на кліренс секукінумабу.
Вплив маси тіла на фармакокінетику
Кліренс секукінумабу та об’єм розподілу збільшуються зі збільшенням маси тіла.
Діти
У двох педіатричних дослідженнях пацієнти із помірним або тяжким бляшковим псоріазом (віком від 6 до 18 років) застосовували секукінумаб в рекомендованих для дітей дозах. На 24 тижні у пацієнтів із масою тіла ≥ 25 та < 50 кг середня мінімальна концентрація ± СВ в рівноважному стані становила 19,8 ± 6,96 мкг/мл (n = 24) після прийому 75 мг секукінумабу, а у пацієнтів із масою тіла ≥ 50 кг середня концентрація ± СВ становила 27,3 ± 10,1 мкг/мл (n = 36) після прийому 150 мг секукінумабу. Середня мінімальна концентрація ± СВ в рівноважному стані у пацієнтів з масою тіла < 25 кг (n = 8) становила 32,6 ± 10,8 мкг/мл на 24 тижні після прийому дози 75 мг.
Клінічні характеристики
Скафо Показання
Бляшковий псоріаз у дорослих.
Лікарський засіб Скафо призначений для лікування бляшкового псоріазу середньотяжкого або тяжкого перебігу у дорослих пацієнтів, яким показана системна терапія.
Бляшковий псоріаз у дітей
Лікарський засіб Скафо призначений для лікування бляшкового псоріазу середньотяжкого або тяжкого перебігу у дітей та підлітків віком від 6 років, яким показана системна терапія.
Псоріатичний артрит.
Лікарський засіб Скафо як монотерапія або в комбінації з метотрексатом призначений для лікування активного псоріатичного артриту у дорослих пацієнтів при недостатній відповіді на попередню терапію протиревматичними препаратами, що змінюють перебіг захворювання (DMARD) (див. розділ «Фармакодинаміка»).
Аксіальний спондилоартрит
Анкілозуючий спондиліт (АС, рентгенологічний аксіальний спондилоартрит).
Лікарський засіб Скафо призначений для лікування активного анкілозуючого спондиліту у дорослих пацієнтів при недостатній відповіді на стандартну терапію.
Нерентгенологічний аксіальний спондилоартрит
Лікарський засіб Скафо призначений для лікування активного нерентгенологічного аксіального спондилоартриту з об’єктивними ознаками запалення, про що свідчить підвищений рівень С-реактивного білка (СРБ) та/або магнітно-резонансна томографія (МРТ), у дорослих, які неналежним чином реагували на нестероїдні протизапальні препарати (НПЗП).
Протипоказання
Тяжкі реакції гіперчутливості до діючої речовини або до будь-якого допоміжного компонента препарату, що наведені у розділі «Склад».
Клінічно значущі інфекції в стадії загострення (наприклад активний туберкульоз; див. розділ «Особливості застосування»).
Особливі заходи безпеки.
Одноразовий флакон містить 150 мг секукінумабу для відновлення стерильною водою для ін’єкцій. Отриманий розчин має бути прозорим та безбарвним або світло-жовтого кольору. Не використовуйте, якщо ліофілізований порошок не повністю розчинився або якщо рідина містить чітко видимі частинки, каламутна або має помітно коричневий колір.
Відновлення
Лікарський засіб Скафо, порошок для розчину для ін’єкцій по 150 мг, повинен готувати медичний працівник. Приготування розчину для підшкірної ін’єкції має здійснюватися без перерв і в асептичних умовах. Час приготування від проколювання пробки до кінця відновлення становить в середньому 20 хвилин і не має перевищувати 90 хвилин.
1. Флакон з порошком довести до кімнатної температури та переконатися в тому, що стерильна вода для ін’єкцій досягла кімнатної температури.
2. Відібрати в одноразовий шприц об’ємом 1 мл рівно 1,0 мл стерильної води для ін’єкцій.
3. Зняти пластикову кришку з флакона.
4. Голку шприца вставити у флакон, що містить порошок, через центр гумової насадки на флаконі та відновити порошок шляхом повільного введення 1,0 мл стерильної води для ін’єкцій у флакон. Струмінь стерильної води для ін’єкцій має бути спрямований у центр порошку.
5. Нахилити флакон під кутом приблизно 45° та обережно обертати між пальцями протягом близько 1 хвилини. Флакон не струшувати та не перевертати.
6. Флакон витримати при кімнатній температурі протягом щонайменше 10 хвилин до повного розчинення порошку. При приготуванні розчину може утворитись піна.
7. Після цього повторно нахилити флакон під кутом приблизно 45° та обережно обертати між пальцями протягом близько 1 хвилини. Флакон не струшувати та не перевертати.
8. Флакон витримати при кімнатній температурі протягом ще приблизно 5 хвилин. Одержаний розчин має бути прозорий. Його колір може змінюватись від прозорого до світло-жовтого. Не використовувати, якщо ліофілізований порошок повністю не розчинився або якщо рідина містить легко видимі частки, каламутна або має коричневе забарвлення.
9. Готують необхідну кількість флаконів (2 флакони для дози 300 мг).
Будь-який невикористаний лікарський засіб чи відходи потрібно утилізувати відповідно до місцевих вимог.
Застосування дітям
Для дітей, які отримують дозу 75 мг, наразі рекомендується використовувати одноразовий флакон, що містить 150 мг секукінумабу для відновлення у стерильній воді для ін’єкцій. Слід взяти трохи більше 0,5 мл відновленого розчину для підшкірних ін’єкцій, а залишки розчину слід одразу утилізувати.
Взаємодія з іншими лікарськими засобами та інші види взаємодій
На тлі терапії лікарським засобом Скафо не слід проводити вакцинацію живими вакцинами (див. також розділ «Особливості застосування»).
Під час дослідження за участю пацієнтів із бляшковим псоріазом жодної взаємодії секукінумабу з мідазоламом не спостерігалось (субстрат CYP3A4).
Під час досліджень артриту жодної взаємодії не спостерігалось при застосуванні секукінумабу одночасно з метотрексатом та/або кортикостероїдами (зокрема у пацієнтів із псоріатичним артритом та аксіальним спондилоартритом).
Особливості застосування
Відстежуваність
З метою покращення відстежуваності біологічних лікарських засобів назву та номер серії застосованого лікарського засобу потрібно чітко записувати.
Інфекції
Секукінумаб може збільшувати ризик розвитку інфекцій. В післяреєстраційний період у пацієнтів, які отримували секукінумаб, спостерігались серйозні інфекції. Слід дотримуватися обережності при вирішенні питання про застосування секукінумабу пацієнтам із хронічною інфекцією або з рецидивуючою інфекцією в анамнезі.
Слід інформувати пацієнта про необхідність звернення до лікаря у разі появи ознак і симптомів, що дають змогу запідозрити розвиток інфекції. При розвитку серйозної інфекції необхідно ретельно спостерігати за станом пацієнта; терапію секукінумабом слід відкласти аж до припинення інфекційного процесу.
Під час клінічних досліджень у пацієнтів, які отримували секукінумаб, спостерігались інфекції (див. розділ «Побічні реакції»). Більшість з них були інфекціями верхніх дихальних шляхів слабкого або помірного ступеня тяжкості, такими як назофарингіт, та не вимагали припинення лікування.
Зважаючи на механізм дії секукінумабу, під час клінічних досліджень псоріазу про легкі шкірно-слизові кандидозні інфекції частіше повідомлялось в групі застосування секукінумабу, ніж в групі плацебо (3,55 випадку на 100 пацієнто-років при застосуванні секукінумабу в дозі 300 мг проти 1,00 випадку на 100 пацієнто-років при прийомі плацебо) (див. розділ «Побічні реакції»).
У клінічних дослідженнях не отримано повідомлень про збільшення сприйнятливості до туберкульозу, однак секукінумаб не слід застосовувати пацієнтам з активною формою туберкульозу. Перед початком лікування препаратом у пацієнтів з латентною формою туберкульозу слід розглянути можливість проведення протитуберкульозної терапії.
Запальне захворювання кишечнику (в тому числі хвороба Крона та виразковий коліт)
При прийомі секукінумабу повідомлялось про нові випадки або загострення запального захворювання кишечнику (див. розділ «Побічні реакції»). Секукінумаб не рекомендується пацієнтам із запальним захворюванням кишечнику. Якщо у пацієнта з’являються ознаки та симптоми запального захворювання кишечнику або він відчуває загострення існуючого запального захворювання кишечнику, прийом секукінумабу слід припинити та розпочати альтернативну терапію.
Реакції гіперчутливості
Під час клінічних досліджень спостерігались рідкі випадки анафілактичних реакцій у пацієнтів, які отримували секукінумаб. У разі розвитку анафілактичної або іншої алергічної реакції прийом секукінумабу слід негайно припинити та призначити відповідну терапію.
Вакцинація
На тлі терапії секукінумабом не слід проводити вакцинацію живими вакцинами.
Для пацієнта, який отримує терапію секукінумабом, слід застосовувати інактивовані або неживі вакцини. У клінічному дослідженні у схожої кількості здорових добровольців, які отримували 150 мг секукінумабу і плацебо, відзначалась адекватна імунна відповідь після вакцинації менінгококовимита інактивованими протигрипозними вакцинами, що проявлялося у вигляді 4-разового збільшення титру антитіл до вказаних вакцин. Ці дані свідчать про те, що секукінумаб не пригнічує гуморальну імунну відповідь на менінгококові та інактивовані протигрипознівакцини.
Перед початком лікування лікарським засобом Скафо діти мають отримати всі щеплення за віком відповідно до діючих настанов з імунізації.
Супутня імуносупресивна терапія
Під час досліджень псоріазу безпеку та ефективність застосування секукінумабу в комбінації з імуносупресантами, включаючи біологічні препарати чи фототерапію, не вивчали. Під час досліджень артриту (включаючи дослідження за участю пацієнтів із псоріатичним артритом та анкілозуючим спондилітом) секукінумаб одночасно застосовувався з метотрексатом, сульфасалазином та/або кортикостероїдами. Слід дотримуватися обережності при одночасному застосуванні імуносупресантів із секукінумабом (див. розділ «Взаємодія з іншими лікарськими засобами та інші види взаємодій»).
Застосування у період вагітності або годування груддю
Жінки репродуктивного віку
Жінки репродуктивного віку повинні використовувати ефективні методи контрацепції під час лікування та протягом щонайменше 20 тижнів після нього.
Вагітність
Немає достатніх даних щодо застосування секукінумабу вагітним жінкам. Як запобіжний захід під час вагітності лікарський засіб Скафо застосовувати не слід.
Годування груддю
Невідомо, чи проникає секукінумаб у грудне молоко людини. Імуноглобуліни проникають в грудне молоко людини; невідомо, чи всмоктується секукінумаб системно після прийому. Через можливість розвитку побічних реакцій у грудних дітей внаслідок застосування секукінумабу необхідно прийняти рішення про припинення грудного вигодовування під час лікування та щонайменше протягом 20 тижнів після прийому препарату або про припинення терапії лікарським засобом Скафо з урахуванням користі від грудного вигодовування для дитини та переваги терапії для жінки.
Репродуктивна функція
Вплив секукінумабу на репродуктивну функцію для людини не вивчався. Дослідження на тваринах не виявили прямого чи опосередкованого токсичного впливу на репродуктивну функцію.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами
Лікарський засіб Скафо не чинить або чинить незначний вплив на здатність керувати транспортними засобами або працювати з іншими автоматизованими системами.
Спосіб застосування Скафо та дози
Лікарський засіб Скафо призначений для застосування під медичним наглядом за рекомендацією лікаря, який має досвід діагностики та лікування станів, для яких показаний лікарський засіб Скафо.
Дози
Бляшковий псоріаз у дорослих
Рекомендована доза становить 300 мг секукінумабу у вигляді підшкірної ін’єкції як початкова доза на 0, 1, 2, 3 тижні з наступним щомісячним введенням як підтримуючої дози, починаючи з 4 тижня. Кожну дозу 300 мг вводять у вигляді двох окремих підшкірних ін’єкцій по 150 мг.
Бляшковий псоріазу дітей віком від 6 років
Рекомендована доза розраховується за масою тіла (таблиця 7) та вводиться у вигляді підшкірної ін’єкції як початкова доза на 0, 1, 2, 3 тижні з наступним щомісячним введенням як підтримуючої дози, починаючи з 4 тижня. Кожну дозу 75 мг вводять у вигляді однієї підшкірної ін’єкції по 75 мг. Кожну дозу 150 мг вводять у вигляді однієї підшкірної ін’єкції по 150 мг. Кожну дозу 300 мг вводять у вигляді двох підшкірних ін’єкцій по 150 мг.
Таблиця 7
Рекомендована доза для лікуваннябляшкового псоріазу у дітей
Маса тіла пацієнта | Рекомендована доза |
< 25 кг | 75 мг |
Від 25 до < 50 кг | 75 мг |
≥ 50 кг | 150 мг (*може бути збільшена до 300 мг) |
*Деякі пацієнти можуть отримати додаткову користь від прийому підвищеної дози.
Псоріатичний артрит
Для пацієнтів з бляшковим псоріазом помірного або тяжкого ступеня або пацієнтів з неадекватною відповіддю на терапію інгібіторами анти‑ФНПα рекомендована доза становить 300 мг у вигляді підшкірної ін’єкції як початкова доза на 0, 1, 2, 3 тижні з наступним щомісячним введенням як підтримуючої дози, починаючи з 4 тижня. Кожну дозу
300 мг вводять у вигляді двох окремих підшкірних ін’єкцій по 150 мг.
Для інших пацієнтів рекомендована доза становить 150 мг у вигляді підшкірної ін’єкції як початкова доза на 0, 1, 2, 3 тижні з наступним щомісячним введенням як підтримуючої дози, починаючи з 4 тижня. Залежно від клінічної відповіді дозу лікарського засобу можна збільшити до 300 мг.
Аксіальний спондилоартрит
Анкілозуючий спондиліт (АС, рентгенологічний аксіальний спондилоартрит)
Рекомендована доза становить 150 мг у вигляді підшкірної ін’єкції ін’єкції як початкова доза на 0, 1, 2, 3 тижні з наступним щомісячним введенням як підтримуючої дози, починаючи з 4 тижня. На підставі клінічної відповіді дозу можна збільшити до 300 мг. Кожну дозу 300 мг вводять у вигляді двох підшкірних ін’єкцій по 150 мг.
Нерентгенологічний аксіальний спондилоартрит
Рекомендована доза становить 150 мг у вигляді підшкірної ін’єкції як початкова доза на 0, 1, 2, 3 тижні з наступним щомісячним введенням як підтримуючої дози, починаючи з 4 тижня.
Для всіх перерахованих вище показань наявні дані свідчать про те, що клінічна відповідь зазвичай досягається протягом 16 тижнів лікування. Слід розглянути ймовірність припинення лікування для пацієнтів з неадекватною відповіддю на терапію до 16-го тижня лікування. Деякі пацієнти, які спочатку мають часткову відповідь, згодом можуть показати кращу відповідь при продовженні лікування після 16-го тижня.
Особливі групи пацієнтів
Пацієнти літнього віку (понад 65 років)
Корекція дози не потрібна (див. розділ «Фармакокінетика»).
Порушення функції нирок / порушення функції печінки
Застосування лікарського засобу Скафо цим групам пацієнтів не вивчали. Рекомендацій щодо дозування надати не можна.
Діти
Безпека та ефективність терапії лікарським засобом Скафо для дітей з бляшковим псоріазом віком до 6 років не встановлені.
Безпека та ефективність терапії для дітей (віком до 18 років) не встановлені для інших показань. Дані відсутні.
Спосіб застосування
Лікарський засіб Скафо слід вводити у вигляді підшкірної ін’єкції. Якщо можна, місцями ін’єкції не повинні бути ділянки шкіри, уражені псоріазом. Перед використанням порошок для розчину для ін’єкцій має бути відновлений. Відновлення, приготування дози та застосування порошку для приготування розчину для ін’єкцій повинен здійснювати спеціаліст системи охорони здоров’я. Інструкцію з відновлення лікарського засобу перед застосуванням див. у розділі «Особливі заходи безпеки».
Діти
Безпека та ефективність терапії препаратом Скафо для дітей з бляшковим псоріазом віком до 6 років не встановлені.
Безпека та ефективність терапії лікарським засобом Скафо для дітей (віком до 18 років) не встановлені для інших показань. Дані відсутні.
Передозування
Під час клінічних досліджень внутрішньовенно введені дози до 30 мг/кг (приблизно 2000-3000 мг) не супроводжувались дозолімітуючою токсичністю. У разі передозування рекомендується спостереження за пацієнтом для виявлення ознак і симптомів небажаних реакцій. При необхідності слід негайно провести симптоматичне лікування.
Побічні реакції
Огляд профілю безпеки
Найчастішими побічними реакціями є інфекції верхніх дихальних шляхів (найчастіше повідомлялось про назофарингіт, риніт).
Перелік побічних реакцій представлено в таблиці 8.
Побічні реакції, що спостерігались під час клінічних досліджень та протягом післяреєстраційного спостереження (таблиця 8), наведено відповідно до системно-органного класу MedDRA. В межах кожного класу системи органів небажані реакції розподілені за частотою, починаючи з найбільш частих. У кожній групі за частотою небажані реакції на лікарський засіб представлено в порядку зменшення тяжкості. Частоту виникнення небажаних реакцій визначено таким чином: дуже часто (≥ 1/10); часто (від ≥ 1/100 до < 1/10); нечасто (від ≥ 1/1000 до < 1/100); рідко (від ≥ 1/10000 до < 1/1000); дуже рідко (< 1/10000); та невідомо (не можна оцінити на основі наявних даних).
Під час сліпих та відкритих клінічних досліджень за різними показаннями (бляшковий псоріаз, псоріатичний артрит, аксіальний спондилоартрит, анкілозуючий спондиліт та інші аутоімунні захворювання) понад 18000 пацієнтів отримували секукінумаб, що становило 30565 пацієнто-років застосування. При цьому понад 11700 пацієнтів отримували секукінумаб протягом щонайменше одного року. Профіль безпеки секукінумабу однаковий при застосуванні за всіма показаннями.
Таблиця 8 Перелік побічних реакцій, виявлених в клінічних дослідженнях1) та під час післяреєстраційного спостереження
Системно-органний клас | Частота | Побічна реакція |
Інфекції та інвазії | Дуже часто | Інфекції верхніх дихальних шляхів |
Часто | Герпетична інфекція слизової оболонки порожнини рота | |
Мікоз стоп | ||
Нечасто | Кандидозна інфекція порожнини рота | |
Грибкове ураження шкіри стоп | ||
Зовнішній отит | ||
Інфекції нижніх дихальних шляхів | ||
Невідомо | Кандидозна інфекція шкіри і слизових оболонок (включаючи кандидоз стравоходу) | |
Порушення з боку крові та лімфатичної системи | Нечасто | Нейтропенія |
Порушення з боку імунної системи | Рідко | Анафілактичні реакції |
Порушення з боку нервової системи | Часто | Головний біль |
Порушення з боку органів зору | Нечасто | Кон’юнктивіт |
Порушення з боку респіраторної системи, торакальні та медіастинальні порушення | Часто | Ринорея |
Порушення з боку шлунково-кишкового тракту | Часто | Діарея, нудота |
Нечасто | Запальні захворювання кишечнику | |
Порушення з боку шкіри та підшкірної клітковини | Нечасто | Кропив’янка |
Рідко | Ексфоліативний дерматит 2) | |
Загальні порушення та реакції у місці введення | Часто | Втома |
1)Плацебо‑контрольовані клінічні дослідження (фаза III) за участю пацієнтів з бляшковим псоріазом, псоріатичним артритом, анкілозуючим спондилітом та нерентгенологічним аксіальним спондилоартритом, які отримували 300 мг, 150 мг, 75 мг або плацебо протягом періоду до 12 тижнів (псоріаз) чи 16 тижнів (псоріатичний артрит, анкілозуючий спондиліт та нерентгенологічний аксіальний спондилоартрит).
2)Випадки спостерігались у пацієнтів із псоріазом.
Опис окремих побічних реакцій
Інфекції
У плацебо-контрольований період клінічних досліджень бляшкового псоріазу (всього 1382 пацієнти отримували секукінумаб та 694 пацієнти отримували плацебо протягом періоду до 12 тижнів) інфекції були зареєстровані у 28,7 % пацієнтів, які отримували секукінумаб, порівняно з 18,9 % пацієнтів, які отримували плацебо. Більшість інфекцій не були серйозними і включали інфекції верхніх дихальних шляхів легкого або помірного ступеня тяжкості, такі як назофарингіт, і не вимагали припинення лікування. Спостерігалась підвищена частота шкірного кандидозу або кандидозу слизових оболонок, що узгоджувалась із механізмом дії, але випадки були легкими та помірними за ступенем тяжкості, несерйозними, мали стандартну відповідь на лікування і не вимагали припинення лікування. Серйозні інфекції спостерігались у 0,14 % пацієнтів, які отримували секукінумаб, та у 0,3 % пацієнтів, які отримували плацебо (див. розділ «Особливості застосування»).
Протягом усього періоду лікування (загалом 3430 пацієнтів, більшість з яких отримували секукінумаб протягом 52 тижнів) інфекції були зареєстровані у 47,5 % пацієнтів, які отримували секукінумаб (0,9 випадку на пацієнто-рік подальшого спостереження). Серйозні інфекції були зареєстровані у 1,2 % пацієнтів, які отримували секукінумаб (0,015 випадку на пацієнто-рік подальшого спостереження).
Частота інфекцій, що спостерігалась під час проведення клінічних досліджень псоріатичного артриту та аксіального спондилоартриту (анкілозуючого спондиліту та нерентгенологічного аксіального спондилоартриту), була подібною до тієї, що спостерігалась під час досліджень псоріазу.
Нейтропенія
Під час клінічних досліджень псоріазу фази 3 нейтропенія частіше спостерігалась в групі секукінумабу, ніж в групі плацебо, але більшість випадків бути слабкими, минущими та оборотними. Нейтропенія < 1,0-0,5 × 109/л (ступінь 3 за CTCAE) спостерігалась у 18 з 3430 (0,5 %) пацієнтів, які отримували секукінумаб. При цьому відсутність залежності від дози та відсутність тимчасового причинно-наслідкового зв’язку із інфекціями спостерігалась в 15 з 18 випадків. Про більш тяжкі випадки нейтропенії не повідомлялось. В інших трьох випадках повідомлялося про несерйозні інфекції зі звичайною реакцією на стандартну терапію, які не вимагали припинення лікуванням секукінумабом.
Частота нейтропенії при псоріатичному артриті та аксіальному спондилоартриті (анкілозуючому спондиліті та нерентгенологічному аксіальному спондилоартриті) аналогічна частоті, що спостерігається при псоріазі.
Рідко спостерігались випадки нейтропенії < 0,5 × 109/л (ступінь 4 за CTCAE).
Реакції гіперчутливості
Під час клінічних досліджень спостерігались випадки кропив’янки та рідко - анафілактичної реакції на секукінумаб (див. розділ «Особливості застосування»).
Імуногенність
Під час клінічних досліджень псоріазу, псоріатичного артриту та аксіального спондилоартриту (анкілозуючого спондиліту та нерентгенологічного аксіального спондилоартриту) у менше ніж 1% пацієнтів, які отримували секукінумаб, спостерігалось утворення антитіл до секукінумабу до 52 тижня лікування. Близько половини антитіл, що утворилися на тлі терапії, були нейтралізуючими, що не асоціювалось із втратою ефективності терапії або із впливом на фармакокінетичні параметри лікарського засобу.
Діти
Побічні ефекти у дітей віком від 6 років з бляшковим псоріазом
Безпеку секукінумабу вивчали у двох дослідженнях фази III за участю дітей з бляшковим псоріазом. Перше дослідження (педіатричне дослідження 1) було подвійно сліпим, плацебо-контрольованим дослідженням за участю 162 пацієнтів віком від 6 до 18 років з тяжким бляшковим псоріазом. Друге дослідження (педіатричне дослідження 2) було відкритим дослідженням за участю 84 пацієнтів віком від 6 до 18 років з помірним або тяжким бляшковим псоріазом. Профіль безпеки, про який повідомлялося в цих двох дослідженнях, був порівнянним із профілем безпеки, про який повідомлялося у дорослих хворих на бляшковий псоріаз
Повідомлення про підозрювані побічні реакції
Після реєстрації лікарського засобу важливо повідомляти про підозрювані побічні реакції. Це забезпечує постійний моніторинг співвідношення користь/ризик застосування лікарського засобу. Спеціалістів сфери охорони здоров’я просять звітувати про будь-які підозрювані небажані реакції через національну систему звітності (автоматизована інформаційна система з фармаконагляду).
Термін придатності Скафо
3 роки.
Після відновлення.
Хімічна та фізична стабільність приготованого розчину була продемонстрована протягом 24 годин при температурі від 2 до 8 °C.
З мікробіологічної точки зору, якщо метод відновлення не виключає ризику мікробіологічного забруднення, лікарський засіб необхідно використати негайно.
Умови зберігання Скафо
Зберігати при температурі 2-8 °C у недоступному для дітей місці. Не заморожувати.
Упаковка
Флакон з прозорого безбарвного скла, закупорений сірою гумовою пробкою і алюмінієвим ковпачком типу flip-off білого кольору. в коробці з картону пакувального.
Категорія відпуску
За рецептом.
Виробник
Новартіс Фарма ГмбХ/ Novartis Pharma GmbH.
Місцезнаходження виробника
Рунштрассе 25 та Обер Турнштрассе 8, Нюрнберг, 90429, Німеччина/
Roonstrasse 25 und Obere Turnstrasse 8, Nuernberg, 90429, Germany
Подальша інформація
Пам'ятайте, зберігайте ці та всі інші ліки в недоступному для дітей місці, ніколи не передавайте свої ліки іншим і використовуйте Скафо тільки за призначенням.
Завжди консультуйтеся зі своїм лікарем, щоб переконатися, що інформація, яка відображається на цій сторінці, може бути застосована до ваших особистих обставин.
Увага! Ця інструкція для медичного застосування лікарського засобу є офіційною інструкцією виробника Новартіс Фарма Штейн АГ.
Авторське право:
- https://www.novartis.com/ - Новартіс Фарма Штейн АГ
- http://www.drlz.com.ua - Державний реєстр ЛЗ України
| Тип даних | Відомості з реєстру |
| Торгівельне найменування: | Скафо |
| Виробник: | Новартіс Фарма Штейн АГ |
| Форма випуску: | порошок для розчину для ін'єкцій, по 150 мг; по 1 флакону в коробці з картону пакувального |
| Реєстраційне посвідчення: | UA/18114/01/01 |
| Дата початку: | 19.08.2020 |
| Дата закінчення: | 19.08.2025 |
| Міжнародне непатентоване найменування: | Secukinumab |
| Умови відпуску: | за рецептом |
| Склад: | 1 флакон з порошком містить 150 мг секукінумабу |
| Фармакотерапевтична група: | Імуносупресанти, інгібітори інтерлейкіну. |
| Код АТС: | L04AC10 |
| Заявник: | Новартіс Оверсіз Інвестментс АГ |
| Країна заявника: | Швейцарія |
| Адреса заявника: | Ліхтштрассе 35, 4056 Базель, Швейцарія |
| Тип ЛЗ: | Звичайний |
| ЛЗ біологічного походження: | Нi |
| ЛЗ рослинного походження: | Нi |
| Гомеопатичний ЛЗ: | Нi |
| Тип МНН: | Моно |
| Дострокове припинення | Нi |
| Код ATC | Назва групи |
| L | Антинеопластичні та імуномодулюючі засоби |
| L04 | Імунодепресанти |
| L04A | Імунодепресанти |
| L04AC | Інгібітори інтерлейкіну |
| L04AC10 | Cекукінумаб |