Спадкова нефропатія
Етіологія і патогенез
Спадкова нефропатія — дифузне ураження нирок, яке спостерігають у кількох членів однієї сім’ї з домінантним або рецесивним типом успадкування хвороби.
Перше повідомлення про спадкову нефропатію належить W. Dickinson (1881), який виявив гематурію і протеїнурію у кількох членів однієї сім’ї і трактував захворювання як сімейний нефрит.
До групи спадкових нефропатій входять нефритоподібні синдроми, досить схожі за клінічною картиною з набутими захворюваннями нирок — хронічним гломерулонефритом або хронічним пієлонефритом (спадковий нефрит, сімейний ідіопатичний нефронофтиз), — і хронічні тубулопатії, зумовлені генетичним дефектом транспортування органічних сполук і електролітів у ниркових канальцях, унаслідок чого виникають різноманітні обмінні порушення в організмі (нирковий тубулярний ацидоз, фосфат-діабет тощо). Канальцеві синдроми перебігають як рахітоподібні захворювання і їх найчастіше спостерігають у дитячому віці.
Спадковий нефрит. У дорослих з усіх спадкових нефропатій найчастіше діагностують спадковий хронічний нефрит, який може проявлятися лише ураженням нирок або поєднуватися з дефектом слуху.
У 1927 p. A. Alport вказав на зв’язок сімейного нефриту з невритом слухових нервів. За пропозицією D. Williamson (1961), спадковий нефрит, що поєднується з глухотою, отримав назву "синдром Альпорта". У подальшому при спадковому нефриті були виявлені також ураження очей та аномалії кісткового скелета.
Синдром Альпорта (спадковий нефрит) — прогресивне генетично зумовлене порушення гломерулярної фільтрації, що супроводжується еритроцитурією і розвитком хронічної ниркової недостатності. Класично відомий синдром Альпорта має також екстраренальні ознаки, а саме глухоту чи порушення зору, яке частіше проявляється у хворих жіночої статі. У нефрологічній практиці діагноз синдрому Альпорта, як правило, запідозрюють клінічно у віці 5—7 років за наявності еритроцитурії із супутнім високочастотним зниженням слуху та верифікують на підставі результатів нефробіопсії і генетичного тестування.
Проте більшість дослідників вважають за доцільне виділити спадковий нефрит як самостійну нозологічну форму незалежно від наявності патології слуху, а синдром Альпорта розглядають як клінічний варіант спадкового нефриту із прогностично найнесприятливішим перебігом хвороби.
За сучасними даними, синдром Альпорта найчастіше є причиною еритроцитурій у дитячому віці. Це пов’язано насамперед із впровадженням генної діагностики. За даними генетичних досліджень, частота синдрому Альпорта значно перевищує відомий нефрит з туговухістю і сягає 1:5000—10 000 населення. Таким чином, випадково виявлена еритроцитурія у дітей найчастіше є ознакою одного із варіантів синдрому Альпорта.
Захворювання успадковується за автосомно-домінантним типом, частково пов’язаним зі статтю або залежним від статі. Змінений ген локалізується в Х-хромосомі, хворі чоловіки-гетерозиготи можуть передавати захворювання лише донькам, а хворі жінки — більшості доньок і синів. Цим пояснюється тяжчий перебіг хвороби у чоловіків. Проте описані поодинокі випадки передавання захворювання від батька до сина, що не укладається в поняття успадкування, зчепленого зі статтю. Очевидно, мутантний ген здатний до кросинговеру між гомологічними ділянками X- і Y-xpoмосом. Можливість асоціації автосом, що несуть патогенний ген з Х-хромосомою, дає змогу пояснити наявність великої кількості носіїв захворювання в сім’ях зі спадковим нефритом.
Механізм розвитку спадкового нефриту не з’ясований. Є дані, що ураження нирок і очей, неврит слухових нервів при цьому захворюванні — це наслідок дефекту розвитку одного з ланцюгів колагенезу IV на рівні гена.
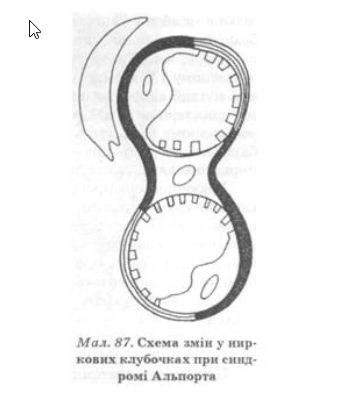
Патогенетичною основою морфологічних змін при захворюванні є дефіцит колагену IV типу в базальних мембранах капілярів клубочка і канальців, що призводить до їх розширення (мал. 87). Подальші зміни спричинюють гіалінізацію клубочків і розвиток ниркової недостатності.
Альфа 1, 2, 5 та 6 містяться в епітеліальній базальній мембрані, альфа 1—5 — у базальній мембрані гломерул. Наявність альфа 5 в обох видах мембран дозволяє проводити діагностику на підставі біопсії шкіри за наявності дефекту при синдромі Альпорта.
Альфа 3(IV)- і альфа 4(ГУ)-ланцюги наявні в гломерулярній та частково тубулярній базальних мембранах. Альфа 5(IV) розподіляється в гломерулярній базальній мембрані, капсулі ниркового клубочка (капсулі Шумлянського— Боумена), частково — у дистальних канальцевих базальних мембранах. Альфа 6(ІУ)-ланцюг наявний у капсулі ниркового клубочка і частково міститься в тубулярній мембрані та на початку розвитку капілярних петель гломерул.
Нормальні гломерулярні капіляри фільтрують плазму крізь базальну мембрану, багату на альфа 3, 4 і 5-ланцюги колагену IV типу. Найчастіше при синдромі Альпорта відсутній альфа5-ланцюг, який заміщується ембріонально незрілими альфа 1 і 2-ланцюгами колагену IV типу та колагенами V і VI типу. їх наявність супроводжується неконтрольованим виробленням протеаз (колагеназ і катепсинів). Останні призводять до розщеплення базальної мембрани за відсутності зміцнювальної дії альфа 5-ланцюга.
За типом успадкування визначають синдром Альпорта домінантний, зчеплений з Х-хромосомою (мутації гена COL4A5), автосомнодомінантний (COL4A3 і COL4A4 хромосоми 2, частіше у чоловіків) та автосомнорецесивний (COL4A3 або COL4A4). Виділяють також XL субтип з дифузним лейоміоматозом та мегакаріоцитарною тромбоцитопенією (мутації генів, які кодують альфа 5 і 6-ланцюги). Усього налічується понад 200 різних мутацій колагену IV типу, які встановлюють шляхом генеалогічного та генетичного аналізів.
Дефекти генотипу визначаються відсутністю (або зменшенням кількості) колагену. Альфа 3, 5-ланцюги колагену IV типу наявні, крім гломерул, у базальній мембрані капсули кришталика, епітеліальній мембрані рогівки і мембрані завитки, базальних мембранах шкіри та легень. Звідси клінічне ураження нирок, очей, вуха та сполучнотканинні стигми дисембріогенезу. При зчепленому з Х-хромосомою синдромі Альпорта (80% всіх випадків) виявляють мутації альфа 5-ланцюга IV типу колагену. При дифузному лейоміоматозі спостерігають майже повну відсутність альфа З—5(IV). При автономно-рецесивному успадкуванні втрачаються альфа 3 і 4-ланцюги, альфа 5 — із базальної гломерулярної мембрани, але альфа 5—6 зберігаються в капсулі ниркового клубочка та дистальній тубулярній мембрані.
Ураження зору проявляються як передній лентиконус у разі ушкодження капсули кришталика, перимакулярні дефекти, або дефекти навколо жовтої плями сітківки (мембрана Bruch), задня поліморфна дистрофія (мембрана Descemet), рецидивна корнеальна ерозія (епітеліальна мембрана рогівки). Глухоту відзначають при ураженні базальної мембрани завитки та непосмугованих м’язових волокон вуха.
Учені висловлюють припущення, що гени мутантів, які спричинюють ураження нирок і органів слуху, розташовані в одній хромосомі, але вони можуть успадковуватися незалежно один від одного. Тому в нащадків пацієнтів, які страждають на глухоту, можуть бути діти з ураженням нирок.
Патологічна анатомія
Патологічні зміни в нирках залежать від стадії, еволюції процесу і статі хворого. За даними біопсії нирок, у ранній період хвороби гістологічні зміни в нирковій тканині відсутні. У пізнішій стадії гістологічна картина характеризується гломерулітом — від фокально-сегментарного, мінімального до дифузного, проліферативно-мембранозного. Фібро- пластичні зміни в клубочках зазвичай відсутні, склеротичні явища виражені різною мірою.
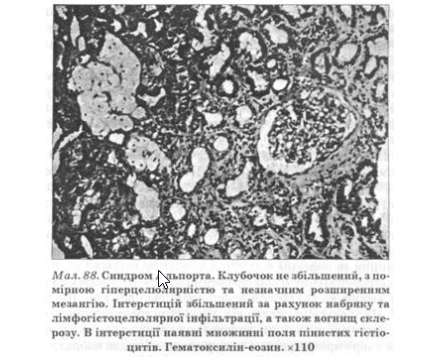
Найтяжчі морфологічні зміни виявляють у чоловіків. Водночас із проліферативним гломерулітом виражені інтерстиціальна проліферація і склероз, а також дистрофія ниркових канальців. Нерідко в інтерстицїї виявляють піняві клітини, що містять нейтральний жир у цитоплазмі і вважаються патогномонічними для синдрому Альпорта (мал. 88). Проте описані клітини спостерігають не в усіх випадках, іноді їх виявляють у хворих з набутими захворюваннями нирок, унаслідок чого їхня специфічність для синдрому Альпорта сьогодні дискутується. Іноді при спадковому нефриті виявляють морфологічну картину, характерну для хронічного пієлонефриту.
У хворих, які померли від прогресивної ниркової недостатності, на автопсії виявляють зменшення розмірів нирок, стоншення кіркової речовини, зморщування і гіаліноз клубочків; у деяких клубочках клітинні інфільтрати мають вигляд півмісяців, виражена тубулярна атрофія, в інтерстиційній тканині — вогнища склерозу і лімфоцитарні інфільтрати, склеротичні зміни судин.
Сучасна клінічна класифікація передбачає наявність ювенільного (розвиток хронічної ниркової недостатності до 30 років) та дорослого типів синдрому Альпорта.
Хворі жіночої статі за наявності зчепленого з Х-хромосомою синдрому Альпорта мають прогностично сприятливий перебіг захворювання. За наявності інших типів успадкування клінічна симптоматика суттєво не залежить від статі. Таким чином, "класичний" синдром Альпорта (гематурійний варіант гломерулонефриту, глухота та розвиток хронічної ниркової недостатності) не єдина форма захворювання, навпаки, цей варіант є малопоширеним. Отже, випадково виявлена гломерулярна еритроцитурія потребує передусім диференціації з генетичними формами синдрому Альпорта. Більше того, навіть за наявності класичного синдрому Альпорта глухота може бути відсутня.
Клінічна картина
Перші ознаки ураження нирок при спадковому нефриті зазвичай виявляють у дитячому віці, найчастіше в період від 3 до 10 років. Початкові симптоми цього захворювання іноді спостерігають у віці до 2 років. Клінічна картина захворювання залежить від статі хворого. У всіх чоловіків захворювання розпочинається з постійної або зворотної макро- чи мікрогематурії, яку може провокувати інфекція верхніх дихальних шляхів. У такому разі хворим помилково ставлять діагноз гострого гломерулонефриту. Зазвичай захворювання прогресує і до 30—40 років розвивається хронічна ниркова недостатність. Розвиток останньої до 16-річного віку не характерний.
У жінок захворювання перебігає в легкій формі і проявляється тільки постійною або зворотною еритроцитурією. У деяких хворих відзначають лейкоцитурію, пов’язану з інфекцією сечових шляхів, яка приєднується. Розвиток ниркової недостатності не характерний. Нерідко в осіб жіночої статі зі стертим перебігом спадкової нефропатії сечовий синдром уперше виявляють у віці 30—40 років під час цілеспрямованого обстеження після виявлення синдрому Альпорта в інших членів сім’ї.
Лабораторно-інструментальні дослідження. Спонтанний характер макрогематурії не виключає ймовірності її виникнення у зв’язку з інтеркурентним захворюванням. У проміжках між періодами макрогематурії зберігається еритроцитурія — від 2 • 106 — 3 • 106 до 100 • 105 в 1 л і більше, яка часто поєднується з протеїнурією.
Кількість білка в загальному аналізі сечі не перевищує 1 г/л і лише в період макрогематурії вона збільшується до 1,65—3,3 г/л. Добова екскреція білка становить 0,5—1,5 г. Протеїнурія, як правило, має непостійний характер.
Лейкоцитурію виявляють рідше, ніж гематурію, але в деяких хворих, переважно жінок, вона буває значнішою, ніж еритроцитурія, а іноді навіть поєднується з вираженою бактеріурією. У таких випадках можна говорити про розвиток пієлонефриту внаслідок вибіркового підвищення чутливості ниркової тканини та інфекції.
Щодо екстраренальних ознак ураження нирок, то набряки і артеріальна гіпертензія у хворих на спадковий нефрит зазвичай відсутні. Лише у хлопчиків віком 7—14 років і у чоловіків при загостренні сечового синдрому з’являються пастозність тканин, артеріальна гіпертензія, яка в подальшому збільшується в міру прогресування захворювання. Інколи набряки і артеріальну гіпертензію спостерігають і в жінок.
Зниження слуху значно частіше виявляють у чоловіків, воно проявляється переважно у віці 8—10 років, а іноді і в пізніші терміни — у період з 11 до 15 років і навіть у 20 і 30 років. У деяких хворих туговухість може бути першим симптомом хвороби.
На ранніх стадіях захворювання зниження слуху виявляють лише при аудіографії, що дає змогу визначити порушення засвоєння звуків з частотою 4099—2011 Гц. Туговухість не супроводжується вестибулярними розладами і, як правило, прогресує. Імовірність прояву ознак невриту слухових нервів у будь-якому віці свідчить про необхідність проведення постійного аудіометричного контролю у хворих на спадковий нефрит.
Ураження очей при синдромі Альпорта трапляється рідше, ніж неврит слухового нерва, проявляється пігментним ретинітом, катарактою, сферофакією, астигматизмом.
Рідшим, ніж глухота, але майже патогномонічним для синдрому Альпорта симптомом є передній лентиконус. На передній поверхні кришталиків спостерігають "олійну краплю", яка може призвести до порушення рефракції і полів зору, заважає огляду очного дна. У хворих із синдромом Альпорта та їхніх родичів на очному дні поблизу щільної плями знаходять також жовтуваті або білі плями.
Для спадкового нефриту характерні ознаки дизембріогенезу: аномалії розвитку нирок і сечових шляхів, уроджені вади серця та інших органів.
Діагностика
Дослідження функціональної здатності нирок у хворих із синдромом Альпорта дає змогу встановити, що у чоловіків уже у відносно ранні терміни захворювання проявляється зниження клубочкової фільтрації за кліренсом ендогенного креатиніну при нормальних величинах, що характеризують парціальні тубулярні функції нирок (екскреція водневих йонів, канальцева реабсорбція води та ін.).
У міру прогресування синдрому Альпорта водночас зі зниженням фільтраційної функції нирок визначають електролітні порушення і метаболічний ацидоз, ступінь вираженості яких залежить від стадії хронічної ниркової недостатності. У хворих жінок ознаки ниркової недостатності можуть бути відсутніми протягом багатьох років.
Діагностичні морфологічні ознаки синдрому Альпорта включають проліферацію мезангіальних клітин і матриксу, стоншення стінок капілярів і тубулоінтерстиціальні зміни, які виявляють не раніше ніж на 5—7-му році життя. У подальшому розвиваються інтерстиціальний фіброз, тубулярна атрофія і дилятація, утворюються піняві клітини (спінена цитоплазма), стоншуються та подвоюються базальні мембрани клубочків і канальців, lamina densa набуває структури, схожої на плетену корзину (див. вклейку, мал. 89). Зі збільшенням тривалості синдрому Альпорта, який визначається генотипом хворого, гломерулосклероз та інтерстиційний фіброз прогресують, розвивається хронічна ниркова недостатність. Імунофлуоресцентні дослідження на ранній стадії не інформативні. Пізніше виявляють гранулярні депозити СЗ-комплементу та імуноглобуліну М. За допомогою імунофлуоресцентних методів дослідження нефробіоптату виявляють дефекти кількості а-ланцюгів колагену, що є патогномонічною ознакою синдрому Альпорта.
Рентгенологічне дослідження дає змогу виявити деякі особливості, характерні для спадкового нефриту. На екскреторних урограмах нерідко відзначають вади розвитку: подвоєння мисок, сечоводів, ротацію нирки, звуження мисково-сечовідного сегмента. Критерії діагнозу синдрому Альпорта: наявність гематурії та порушень слуху (зору — за рахунок стоншення капсули кришталика та переднього лентиконуса у жінок) у родичів чоловічої статі, розвиток захворювання в 7—11-річному віці, розвиток порушень функцій нирок, характерна картина за даними нефробіопсії (мезангіальна проліферація, стоншення стінки капілярів, інтерстиціальний фіброз, тубулярна дистрофія і дилятація, стоншення тубулярної базальної мембрани, наявність пінявих клітин) або біопсії шкіри. Х-хромосомний домінантний синдром Альпорта (COL4A5), автосомнодомінантний і автосомнорецесивний синдром Альпорта (COL4A3 і COL4A4): рентгенологічні зміни чашечково-мискової системи зазвичай мають "пієлонефритичний" характер — асиметрія заповнення мисок, їх дилатація, огрубіння зводів, каліектазія тощо. У деяких хворих екскреторна урографія не виявляє патологічних змін.
При цистографії в окремих спостереженнях виявляють міхурово-сечовідний рефлюкс. Обструктивні уропатїї частіше спостерігають при синдромі Альпорта.
Діагностика.Синдром Альпорта слід підозрювати тоді, коли наявні такі симптоми, як еритроцитурія, ниркова недостатність і зниження (втрата) слуху. Особливо це стосується тих хворих, у родичів яких спостерігались випадки глухоти або захворювання нирок. Крім того, варто пам’ятати, що еритроцитурія є головним проявом синдрому Альпорта і виникає на початку захворювання, у дитячому або юнацькому віці. Тому диференціальний діагноз слід проводити з усіма захворюваннями, що супроводжуються еритроцитурією. Однак частіше синдром Альпорта доводиться диференціювати з доброякісною сімейною гематурією та гломерулонефритом з відкладенням IgA в клубочках (IgA-нефропатія, хвороба Берже). У табл. 20 наведені окремі симптоми, які можна використати для диференціального діагнозу з цими захворюваннями, що найчастіше є причиною оборотної еритроцитурії.

Перебіг і наслідки спадкового нефриту значною мірою залежать від статі хворого і наявності невриту слухових нервів. У чоловіків захворювання розпочинається рано і неухильно прогресує. Протягом кількох років з моменту виявлення перших ознак захворювання спостерігають лише сечовий синдром і туговухість. У подальші роки з’являються набряки, артеріальна гіпертензія, розвивається глухота. Хворі вмирають від хронічної ниркової недостатності у віці 15—30 років.
За відсутності невриту слухових нервів і ураження очей період компенсації у чоловіків триваліший. Під впливом інтеркурентних інфекцій і фізичного навантаження гематурія зазвичай посилюється незалежно від статі хворого.
Перебіг захворювання у жінок найчастіше доброякісний. Перші симптоми хвороби виникають у більш пізньому віці, ніж у чоловіків, проявляються ізольованим сечовим синдромом, порушення слуху можуть бути відсутніми або тривалий час визначатися лише за допомогою аудіометричної апаратури. У період вагітності у хворих жінок може посилюватися протеїнурія, з’являються набряки, артеріальна гіпертензія, іноді розвивається ниркова недостатність. Після пологів екстраренальні ознаки ураження нирок і ознаки ниркової недостатності зазвичай зникають. Однак не виключена ймовірність у подальшому прогресування захворювання з летальним наслідком.
Диференціальна діагностика спадкового нефриту і набутих дифузних захворювань нирок має практичне значення з огляду на відмінність терапевтичної тактики і прогнозу захворювання.
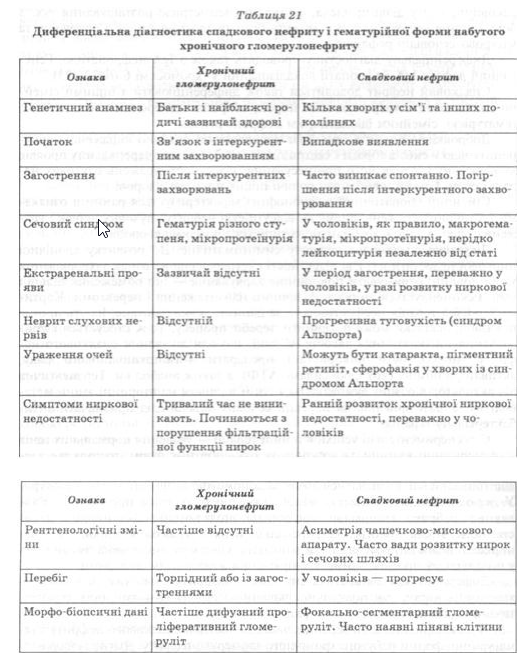
Найскладнішою є диференціальна діагностика спадкового нефриту і гематурійної форми набутого хронічного гломерулонефриту. Діагноз спадкового нефриту встановлюють на підставі даних генетичного анамнезу, спонтанного виникнення і зникнення макрогематурії, наявності невриту слухових нервів, прогресивного характеру захворювання у чоловіків і доброякісного його перебігу в жінок та інших ознак (табл. 21).
Під час проведення диференціальної діагностики слід пам’ятати про синдром брахіо-оторенальний (БОР-синдром), який є автосомнодомінантним і характеризується втратою слуху, прогресивним розвитком хронічної ниркової недостатності та наявністю двобічних вдавлень у ділянці вушної раковини, кіст у ділянці плеча, фістул або асиметрією розташування вух. З боку органів сечової системи можуть бути подвоєння нирок і сечоводів та міхурово-сечовідні рефлюкси.
Диференціальну діагностику проводять також з IgA-нефропатією. Генетичний дефект IgA-нефропатії локалізований у хромосомі 6 (6q22—23).
Спадковий нефрит доводиться також диференціювати з іншими сімейно-спадковими захворюваннями нирок, зокрема з доброякісною сімейною гематурією, сімейним ідіопатичним нефронофтизом.
Доброякісна сімейна гематурія проявляється помірно вираженою еритроцитурією у сибсів (брати і сестри однієї сім’ї) без екстраренальних проявів захворювання, ознак його прогресування, відсутності уражень слухових нервів і очей. Батьки, найближчі родичі інших поколінь здорові.
Сімейний ідіопатичний нефронофтиз характеризується ранніми ознаками тубулярних уражень, анемією, відсутністю невриту слухових нервів і ураження очей, а також домінантного типу успадкування хвороби.
Лікування спадкового нефриту симптоматичне. До розвитку хронічної ниркової недостатності і за відсутності екстраренальних ознак захворювання хворі повинні отримувати повноцінне харчування — без обмеження білків і солі. Рекомендується уникати фізичного навантаження і перевтоми. Кортикостероїдна терапія неефективна, а за даними деяких авторів, навіть протипоказана, оскільки може погіршити перебіг процесу. Те ж стосується і призначення цитостатичних засобів. Є дані, що для лікування спадкового нефриту можуть бути використані препарати 4-амінохінолінового ряду, активатори обміну (кокарбоксилаза, АТФ), а також анаболіки. Терапевтична дія активаторів обміну зумовлюється їхнім впливом на вторинні зміни метаболізму. У разі приєднання інфекції сечових шляхів слід призначати антибактеріальну терапію.
Є експериментальні успіхи в генній терапії — уведення нормальних генів у гломерулярні клітини за допомогою транспортних форм, наприклад аденовірусних капсидів (векторна терапія). Перспективним вважають уведення нормальних генів антенатально (специфічна генна терапія).
Трансплантація нирки є дуже ефективною. Антигломерулярний нефрит у пересадженій нирці формується менше ніж у 5% пацієнтів.
Хворим на спадковий нефрит показана санація вогнищ хронічної інфекції. За наявності хронічного декомпенсованого тонзиліту радикальне лікування доцільне, незважаючи на гематурію. Тонзилектомія протипоказана хворим з ознаками хронічної ниркової недостатності.
При розвитку хронічної ниркової недостатності у міру її прогресування призначають такі ж заходи, як при нирковій недостатності іншої етіології.
Профілактика спадкового нефриту полягає в роз’ясненні хворим ступеня вірогідності ризику захворювання потомства. Медико-генетичної консультації особливо потребують хворі жінки, оскільки матері передають захворювання дітям.
Прогноз
Прогноз захворювання у жінок сприятливий. У чоловіків найчастіше розвивається хронічна ниркова недостатність. Вона може виникати у різному віці, у різних родичів. Розрізняють ювенільну форму захворювання, за якої у дітей розвивається ниркова недостатність у віці до 18 років, і "тип дорослих", за якого ниркова недостатність з’являється у чоловіків віком близько 40 років. Швидкого перебігу хвороби і несприятливого прогнозу слід чекати у хворих, які успадкували захворювання від матері і в яких виявляють протеїнурію та значне загрубіння і розшарування базальних мембран капілярів клубочків.
Подальша інформація
Завжди консультуйтеся зі своїм лікарем, щоб переконатися, що інформація, яка відображається на цій сторінці, може бути застосована до ваших особистих обставин. Інформація призначена тільки для медичних фахівців.