Сімейний ідіопатичний нефронофтиз
Клінічна картина
Сімейний ідіопатичний нефронофтиз — захворювання, що характеризується вираженими дегенеративними і атрофічними змінами в ниркових канальцях, особливо в їхніх дистальних відділах, що призводить до прогресивного фіброзу і швидкого розвитку ниркової недостатності. Належить до сімейно-спадкових нефропатій з рецесивним типом успадкування. Цю патологію ще називають ювенільним нефронофтизом, або хворобою Фанконі. Це автосомно-рецесивна тубулоінтерстиціальна кістозна дисплазія, що не пов’язана зі статтю і ген якої NPHP розташований у довгому плечі 3q21 — q22 хромосоми. Морфологічним субстратом захворювання є нерегулярне стоншення базальних мембран, канальцева атрофія і дилятація, тубулоінтерстиційний склероз та наявність кіст переважно в кортикомедулярній та мозковій зонах, які ушкоджують дистальні звивисті канальці, петлю нефрона і збірні трубочки.
Фанконі зі співавторами в 1951 р. вперше описав своєрідне захворювання нирок у кількох дітей, що належали до двох сімей, не пов’язаних між собою родинними зв’язками. Дані автопсії і прижиттєвої пункції нирок показали, що при нефронофтизі найраніше і найзначніше уражаються канальці, особливо їхні дистальні відділи. У них виявляють виражені дегенеративні та атрофічні зміни. Загибель і розсмоктування ниркової паренхіми спричинюють вторинні запальні зміни в сполучній тканині, що закінчуються прогресивним фіброзом. Клубочки спочатку нормальні, а в стадії вираженого фіброзу запустівають і гіалінізуються. У хворих на нефронофтиз виявляють кістозні зміни в петлі нефрона, які мають істотне значення в розвитку ниркової недостатності.
Безпосередні причини, що призводять до змін канальців, не виявлені. Фанконі висловив припущення, що нефронофтиз пов’язаний з передчасним "зношуванням" нефронів. При нефронофтизі первинним є вроджений дефект ензимних систем епітелію дистальних канальців з подальшим розвитком вторинних змін у проксимальних канальцях і клубочках.
Особи обох статей захворюють однаково часто. Батьки хворих переважно здорові. Найчастіше захворювання носить сімейний характер, але можуть бути і спорадичні випадки.
Клінічна картина. Захворювання починається, як правило, із синдрому нецукрового сечовиснаження, резистентного до пітуїтрину, не раніше ніж через рік після народження, а нерідко у віці 7—8 років або й пізніше. Поліурія супроводжується зниженням відносної густини сечі до 1,006—1,010, у разі обмеження вживання рідини вона може призвести до гострої дегідратації, що проявляється раптовим нез’ясовним підвищенням температури тіла, диспепсичними розладами, судомними скороченнями окремих груп м’язів. Поліурія спочатку не супроводжується протеїнурією, остання з’являється нерідко через кілька років і зазвичай не досягає високого ступеня. Набряки відсутні, артеріальний тиск нормальний і може підвищуватися лише в термінальній стадії. Другим за частотою симптомом є анемія, як правило, нормохромна. Досить часто у хворих на нефронофтиз виявляють ознаки ниркової остеопатії (остеопороз, розширення епіфізів, аномалія скелета, порушення ходи) і відставання у фізичному розвитку.
Наявність нормоклітинної нормохромної анемії пояснюють зниженням еритропоетичної функції нирок. Для нефронофтизу характерні гіпонатріємія, гіпокальціємія, гіпокаліємія, тенденція до зниження артеріального тиску, метаболічний ацидоз. Осмолярність плазми відповідає осмолярності сечі, відносна густина знижена. Уже в молодшому шкільному віці виявляють остеодистрофію та вторинний гіперпаратиреоїдизм. Хворі відстають у фізичному розвитку і в пропорційній будові тіла, але майже не відстають у розумовому розвитку.
Залежно від ураженого локусу гена виділяють три генетичні і фенотипічні варіанти. Інфантильний (дитячий) нефронофтиз (NPH2) характеризується ушкодженням у 9q22—q31 локусі і клінічно маніфестує розвитком хронічної ниркової недостатності у віці до 3 років. У дітей знаходять збільшені нирки з підвищеною ехогенністю і втратою кортикомедулярної диференціації. Клінічно виявляють анемію, гіперкаліємічний метаболічний ацидоз і артеріальну гіпертензію. Морфологічна картина характеризується дифузним склерозивним тубулоінтерстиційним нефритом з мікроцистною дилатацією проксимальних трубочок і капсули ниркового клубочка, наявністю мікроцист у кортикальному шарі нирок (на відміну від медулярних цист у старшому віці при NPH1 і NPH3).
У разі сімейного ідіопатичного нефронофтизу насамперед виявляють гіпо- та ізостенурію, потім установлюють порушення амоніогенезу. Зниження клубочкової фільтрації виявляють пізніше. Рівень натрію і калію в крові зазвичай нормальний. Уміст калію підвищується лише в пізніх стадіях захворювання, при формуванні вираженої ниркової недостатності, коли значно збільшується рівень креатиніну і сечовини. Досить часто і у відносно ранні терміни хвороби спостерігають гіпокальціємію і гіперфосфатемію.
Перебіг хвороби при нефронофтизі неухильно прогресує і призводить до хронічної ниркової недостатності, яка іноді розвивається порівняно швидко (через 2—3 роки). Зазвичай хворі доживають до юнацького віку. Тривалість життя хворих на нефронофтиз з моменту появи перших ознак хвороби становить у середньому 6 років, максимальний термін — 22 роки.
Діагностика
У початковий період діагноз встановити важко через схожість клінічної картини нефронофтизу і нецукрового сечовиснаження, тому захворювання зазвичай розпізнають у стадії ниркової недостатності.
У перебігу нефронофтизу виділяють дві стадії: 1) ізольоване ураження концентраційної функції нирок, що проявляється синдромом ниркового діабету; 2) тотальна ниркова недостатність різного ступеня вираженості. Такий розподіл зумовлює потребу нефрологічного обстеження в кожному випадку пітуїтринрезистентного нецукрового діабету.
У разі поєднання нефронофтизу з мікробно-запальним процесом у нирковій тканині захворювання часто діагностують як хронічний пієлонефрит. Особливо складна диференціальна діагностика нефронофтизу і пієлонефриту при мінімальних проявах сечового синдрому, що поєднується зі зниженням концентраційної здатності нирок за відсутності порушень показників, які характеризують функцію нирок щодо виведення продуктів білкового обміну (табл. 22).
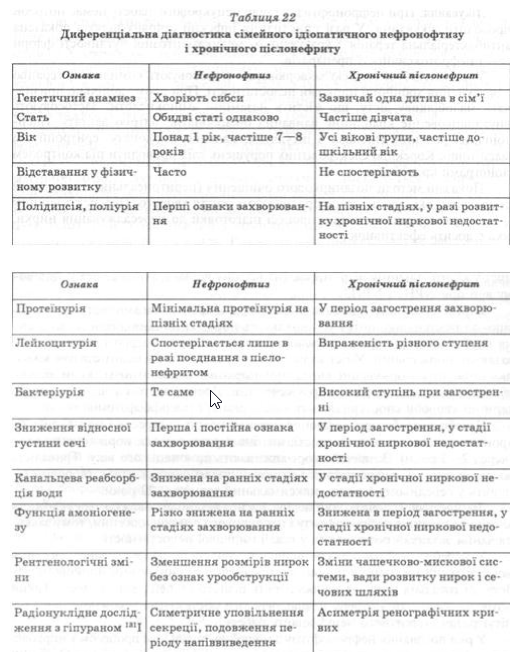
Отже, установлюючи діагноз нефронофтизу, враховують сімейний анамнез, прогресивний перебіг захворювання незалежно від статі хворих, резистентність синдрому нецукрового сечовиснаження до пітуїтрину, наявність протеїнурії та анемії.
Лікування
При нефронофтизі у стадії нецукрового діабету немає потреби проводити лікування. У разі приєднання інфекції сечових шляхів показана антибактеріальна терапія з урахуванням медикаментозної чутливості флори сечі і нефротоксичності препаратів.
У пацієнтів із II стадією захворювання застосовують комплексну терапію для усунення хронічної ниркової недостатності. При цьому доцільно призначати малобілкову дієту, що містить незамінні амінокислоти. Застосовують внутрішньовенне краплинне вливання бікарбонатів, натрію лактату, гіпертонічних розчинів глюкози з інсуліном, кальцію глюконату, еритроцитної маси тощо. Корекцію електролітних порушень слід проводити під контролем йонограми крові і сечі.
Показані методи позаниркового очищення (перитонеальний і хронічний гемодіаліз), які, однак, лише продовжують життя хворого. Перспективнішим є застосування цих методів у процесі підготовки до пересаджування нирки, яка є досить ефективною.
Подальша інформація
Завжди консультуйтеся зі своїм лікарем, щоб переконатися, що інформація, яка відображається на цій сторінці, може бути застосована до ваших особистих обставин. Інформація призначена тільки для медичних фахівців.