Дерматоміозит і поліміозит
Епідеміологія та етіологія
Дерматоміозит — прогресивне генералізоване запальне захворювання з переважним ураженням посмугованих і непосмугованих м’язів і порушенням рухової функції, ураженням шкіри і, часто, внутрішніх органів. За відсутності шкірних уражень (30% хворих) застосовують термін "поліміозит".
Дерматоміозит і поліміозит належать до фупи ідіопатичних запальних міопатій, до якої також входять ювенільний дерматоміозит; міозит, асоційований із системними захворюваннями сполучної тканини — перехресний (overlap) синдром; міозит, асоційований з пухлинами (вторинний); міозит із внутрішньоклітинними включеннями та деякі інші запальні міопатії, що трапляються рідше. Частка дерматоміозиту, поліміозиту і паранеопластично- го міозиту серед ідіопатичних запальних міопатій становить близько 80%.
Епідеміологія. Захворюваність на дерматоміозит/поліміозит коливається в межах 2—10 випадків на 1 млн населення за рік. Пік захворюваності припадає на лютий—квітень і жовтень—листопад. На дерматоміозит і поліміозит хворіють переважно особи віком понад 40 років, співвідношення жінок і чоловіків для дерматоміозиту — 3:1, для поліміозиту — 2:1. Розвиток міозиту, асоційованого з пухлинами, характерний для віку 55—60 років, дещо частіше його діагностують у чоловіків.
Етіологія невідома. Обговорюється тригерна роль вірусної інфекції, насамперед ECHO- та Коксакі-вірусів. Такі припущення базуються на здатності цих вірусів спричинювати у лабораторних тварин патологію м’язів, яка нагадує поліміозит, та виявленні підвищених титрів антитіл до вірусу Коксакі у дітей з дерматоміозитом. Весняний і осінній піки захворюваності на по- ліміозит/дерматоміозит пов’язують саме із сезонністю певних типів вірусних інфекцій. Не виключається значення бактеріальної та паразитарної інфекції. Про роль генетичної схильності свідчить той факт, що поліміозит/дермато- міозит частіше розвивається у кровних родичів хворих і монозиготних близнюків. У хворих на дерматоміозит частіше виявляють імуногенетичні маркери HLA-B8 і HLA-DR3, а при міозиті, асоційованому із системними захворюваннями сполучної тканини, — HLA-B14 і HLA-B40. Уважають, що носійство тих чи тих антигенів системи HLA асоціюється не стільки із самим захворюванням, скільки з особливостями імунних порушень, які проявляються в гіперпродукуванні різних типів міозитспецифічних автоантитіл. Наприклад, антитіла до антигену Jo-1 значно частіше виявляють у носіїв HLA- DR3, вироблення антитіл до PM/Scl асоціюється з носійством HLA-DR3 і HLA-DR4, антитіл до Мі-2 — з HLA-DR7 і HLA-DRw53, антитіл до SRP - з HLA-DR5 і HLA-DRw52.
До додаткових чинників ризику відносять фізичні перевантаження, нервові стреси, перегрівання, переохолодження, гіперінсоляцію, вакцинації, що нерідко передують розвитку дерматоміозиту/поліміозиту та загостренням цих захворювань.
Патогенез
Дерматоміозит і поліміозит — автоімунні захворювання. Не виключено, що поштовхом до розвитку патологічного процесу є віруси, які персистують у м’язах чи клітинах імунної системи і на тлі генетично детермінованих імунорегуляторних дефектів провокують розвиток клітинних і гуморальних автоімунних реакцій проти неінфікованих клітин. Зокрема, це може відбуватися за механізмом молекулярної мімікрії, коли антитіла до певних антигенних детермінант вірусів перехресно реагують з антигенами міофібрил.
Про вагоме значення залучення клітинної ланки імунітету свідчить інфільтрація уражених м’язів активованими Т- і В-лімфоцитами, макрофагами. У процесі імунної відповіді на гіпотетичний інфекційний антиген чи автоантиген активовані Т-лімфоцити синтезують цитокіни, зокрема у-інтерферон, який сприяє експресії молекул адгезії на міоцитах і фіксації до них лімфоцитів. Частина ативованих Т-лімфоцитів виявляє власну цитотоксичну активність стосовно міофібрил. Крім того, Т-супресорні (CD8) лімфоцити синтезують цитотоксичні субстанції (перфорин, гранзим), які залучають до цитотоксичних реакцій інші клітини. У складі клітинних інфільтратів при дерматоміозиті переважають Т-лімфоцити з хелперними властивостями (CD4), В-лімфоцити і макрофаги, а при поліміозиті — цитотоксичні Т-супресорні (CD8) лімфоцити.
У 40% хворих знаходять так звані міозитспецифічні антитіла, спрямовані проти цитоплазматичних білків і рибонуклеїнових кислот, які беруть участь у синтезі білка, а також деяких ядерних субстанцій. Загалом ідентифіковано понад 10 міозитспецифічних антитіл. Найчастіше виявляють антитіла до гістидин-тРНК-синтетази (Jo-1), яка каталізує зв’язування амінокислот з відповідною тРНК (25% хворих); часточок сигнального розпізнавання (SRP) — комплексу білків і молекули РНК, відповідального за перенесення синтезованих молекул білка до ендоплазматичної сітки (до 5% хворих); ядерного білкового комплексу (Мі-2), функція якого невідома (близько 10% хворих). При цьому кожний хворий має зазвичай якийсь один різновид антитіл, що асоціюється з певними клінічними особливостями перебігу захворювання і прогнозом.
У хворих на дерматоміозит/поліміозит часом виявляють і неспецифічні для цього захворювання автоантитіла, у тому числі до міозину, міоглобіну, тиреоглобуліну, ендотеліальних клітин, РФ тощо. Хоча патогенетична роль міозитспецифічних антитіл в ураженні м’язової тканини остаточно не доведена, учені вважають, що специфічні і неспецифічні антитіла, утворюючи імунні комплекси, можуть мати значення в розвитку комплементзалежного ушкодження мікросудин і розвитку позам’язових системних проявів.
У більшості хворих в уражених м’язах знаходять характерну хронічну запальну інфільтрацію периваскулярної та інтерстиційної зон навколо міофібрил. Інфільтрати складаються переважно з лімфоцитів, а також містять у невеликій кількості плазмоцити, гістіоцити, нейтрофіли та еозинофіли. При поліміозиті переважає Т-лімфоцитарна інфільтрація ендомізію, при дерматоміозиті — В-лімфоцитарна інфільтрація перимізію. Типовими є некроз м’язових волокон із втратою посмугованості, їхня дегенерація, активний фагоцитоз загиблих клітин. Часом виявляють ознаки регенерації міофібрил у вигляді малих волокон з великими ядрами. У разі тривалого процесу відзначають багатоядерність міофібрил, атрофію м’язових волокон, фіброз ендо- та перимізію. Судинну патологію при дерматоміозиті діагностують значно частіше, ніж при поліміозиті. Вона проявляється сегментарними проліферативними васкулітами з гіперплазією ендотеліальних клітин, стовщенням інтими дрібних судин, склерозом стінки, мікротромбозом. Продуктивно-некротичний васкуліт частіше буває при ювенільному дерматоміозиті.
Патологічні зміни в біоптатах шкіри закономірно знаходять при дерматоміозиті; при поліміозиті їх може не бути. На початковому етапі розвитку дерматоміозиту, особливо в ділянках еритематозної висипки, характерними є набряк папілярного шару дерми, його інфільтрація Т-лімфоцитами і гістіоцитами, проліферація судин дерми, стовщення епідермісу. На пізніх стадіях виявляють атрофію епідермального шару, переважно дегенеративні зміни і фіброз у дермі, судинну дилатацію.
Гістопатологічна картина у м’язах і шкірі хворих на дерматоміозит і поліміозит малоспецифічна. До того ж у 15—20% хворих із явними клініко-лабораторними ознаками захворювання в біоптатах не вдається знайти характерних морфологічних змін.
Класифікація дерматоміозиту і поліміозиту
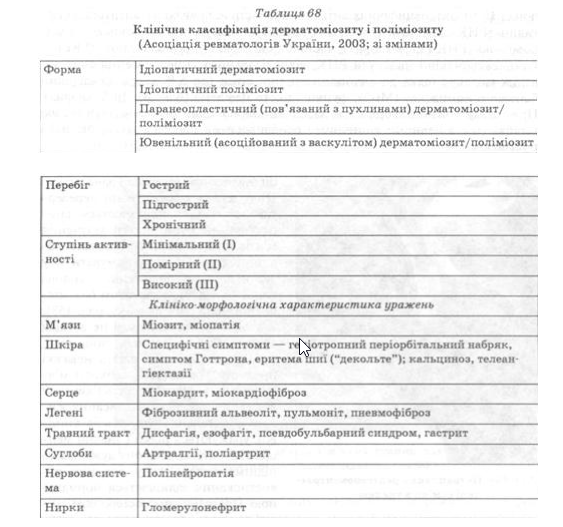
Клінічна картина
У більшості випадків захворювання починається з нездужання, загальної слабкості, поступово з’являється і невпинно прогресує слабкість у проксимальних групах м’язів. Через кілька місяців можлива повна втрата здатності рухатися. У частини хворих розвитку м’язового синдрому передує поява шкірної висипки. Рідше, переважно в осіб молодого віку, спостерігають гострий початок захворювання з гарячкою, болем і слабкістю у м’язах. За кілька тижнів формується розгорнута картина генералізованого ураження м’язів, хворий втрачає масу тіла. Інколи (до 10% хворих) м’язова слабкість прогресує повільно, роками, повної нерухомості не настає, але відбувається атрофія та склероз м’язів, частіше виникають кальцифікати. У 25—30% випадків дебют захворювання характеризується гарячкою, поліміози- том, розвитком поліартралгій чи артритів, специфічної патології шкіри ("руки механіка"), синдрому Рейно і задишки, зумовленої інтерстиційним ураженням легень. Цю групу становлять хворі з особливим клініко-імунологічним субтипом дерматоміозиту/поліміозиту — антисинтетазним синдромом. Його імунологічною ознакою є наявність антитіл до гістидин-тРНК-синтетази (Jo- 1) та інших тРНК-синтетаз, звідки й походить назва синдрому.
Головною клінічною ознакою дерматоміозиту/поліміозиту є симетрична м’язова слабкість у проксимальних групах м’язів верхніх та нижніх кінцівок та м’язах, відповідальних за згинання шиї. Слабкість завжди домінує над міалгіями, які у значної кількості хворих взагалі відсутні.
Хворим важко вставати зі стільця, підніматися сходами, заходити в транспорт, піднімати руки, щоб вдягтися, умитися і зачесатися. Щоб встати з ліжка, вони перевертаються на бік і піднімаються, спираючись на руки. У разі залучення м’язів шиї неможливо відірвати голову від подушки, утримувати її у вертикальному положенні — голова падає на груди — симптом беззаперечної згоди. У тяжких випадках хворі не в змозі ходити без сторонньої допомоги, утримувати в руках навіть неважкі предмети. Унаслідок слабкості м’язів спини та живота можуть з’явитися виражена сутулість, відвисання живота. Дистальні групи м’язів кінцівок уражуються рідко і значно менше. Наприклад, хворому дуже важко піднімати руки вгору, але сила рукостискання залишається нормальною. Проте інколи спостерігають зміну почерку, втрату точності рухів (неможливо повернути ключ у замку, зав’язати шнурки і т.ін.).
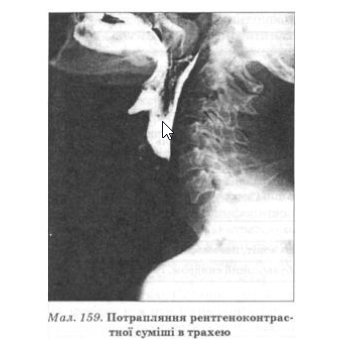
За відсутності своєчасного адекватного лікування до процесу залучаються м’язи глотки і гортані, що призводить до дисфонії (зниження тембру голосу, гугнявість мови) аж до афонії, дизартрії, дисфагії (поперхування при ковтанні). На відміну від ССД, коли проблеми виникають переважно при ковтанні твердої їжі, при поліміозиті утруднене насамперед уживання рідини: унаслідок порушення роботи м’якого піднебіння та надгортанника під час ковтання рідка їжа потрапляє відповідно в ніс і трахею (мал. 159), спричинюючи напад кашлю, витікання їжі через ніс. Оскільки піднебінно-глотковий м’яз-замикач і м’язи верхньої третини стравоходу є посмугованими, вони також залучаються до патологічного процесу, що поглиблює явища дисфагії. Симптоматика, пов’язана з порушенням функції м’язів глотки і гортані з гіпорефлексією м’якого піднебіння, кваліфікується як псевдобульбарний синдром.
При ураженні міжребрових м’язів і діафрагми порушується зовнішнє дихання за рестриктивним типом, знижується життєва ємність легень, що сприяє виникненню пневмоній. У таких хворих на рентгенограмі грудної клітки виявляють двобічну релаксацію куполів діафрагми. В особливо тяжких випадках виникає потреба в інтубації хворого і проведенні штучної вентиляції легень. М’язи обличчя та очей уражаються дуже рідко, проте в деяких хворих може виникати диплопія, двобічний птоз повік.
Під час огляду в половини хворих у гострій фазі захворювання відзначають припухлість, тістувату консистенцію, незначну чи помірну болючість при пальпації уражених м’язів. Шкіра над ними може бути набряклою. Атрофія м’язів на початку хвороби не розвивається. Лише в разі тривалого перебігу дерматоміозиту/поліміозиту внаслідок дистрофії і міолізу м’язових волокон, заміщення їх фіброзною тканиною спостерігають аміотрофії, ущільнення м’язів, виникають сухожилково-м’язові згинальні контрактури. До пізніх ознак належить і кальцифікація м’язів, фасцій, шкіри та підшкірної жирової клітковини, яку легко виявляють рентгенологічно (мал. 160). Кальцинати розташовуються в м’яких тканинах, прилеглих до раніше ушкоджених груп м’язів, тобто переважно в зоні плечового і тазового пояса. Поверхнево розташовані кальцифікати можуть виділятися на поверхню шкіри у вигляді крихтоподібної білої маси. Утворення контрактур і кальцинатів більш типове для ювенільного дерматоміозиту/поліміозиту.
Шкірна висипка трапляється не менше ніж у 2/3 хворих і є ознакою, що відрізняє дерматоміозит від поліміозиту. Типовим варіантом дерматиту є червона чи лілово-пурпурова еритема переважно на відкритих ділянках тіла — обличчі, шиї, передній поверхні грудної клітки, кистях. Завдяки характерному геліотропному (термін походить від назви рослини — геліотропа, колір квітів якого — від яскраво-фіолетового до темно-червоного, пурпурового) відтінку та особливостям локалізації висипка суттєво відрізняється від дерматиту при інших ревматичних хворобах, насамперед СЧВ. Патогномонічними шкірними ознаками дерматоміозиту є періорбітальний набряк з геліотропною еритемою (симптом окулярів) і стійка червоно-пурпурова еритема з лущенням на розгинальних поверхнях п’ястково-фалангових та проксимальних міжфалангових суглобів (симптом Готтрона). У гострий період набряк навколо очей може поширюватись на все обличчя і нагадувати картину набряку Квінке. Часто еритематозна висипка локалізується на лобі, вилицях, крилах носа, носогубних складках, волосистій частині голови, передній поверхні шиї і грудної клітки (зона декольте), розгинальних поверхнях великих суглобів (колінних, ліктьових), верхній частині спини і зовнішній поверхні плечей — симптом шалі (див. вклейку, мал. 163), передньобічних поверхнях стегон. Для хворих на дерматоміозит і поліміозит, які становлять групу пацієнтів з антисинтетазним синдромом, характерним є почервоніння, утворення тріщин та лущення шкіри на подушечках пальців кистей (переважно I, II та III пальців), а також долонь ("руки механіка"). Ця патологія шкіри — єдина, яку спостерігають при поліміозиті.
Рідко при дерматоміозиті трапляються інші варіанти шкірної висипки — папули, пурпура, бульозні висипання, фотодерматит. Незалежно від різновиду ураження шкіри у деяких хворих дерматит може супроводжуватися свербежем.
До типових змін належать також гіперемія і гіпертрофія нігтьових валиків (періунгвальний дерматит), мікроінфаркти (дигітальний артерит) і телеангіектазн по краю нігтьового ложа. Інколи виникають алопеція, посмуго- ваність і ламкість нігтів. У разі тривалого перебігу дерматоміозиту шкіра стає атрофічною, з вогнищами депігментації і пойкілодермїї (чергування вогнищ гіперпігментації та депігментації з множинними телеангіектазіями, сухістю і гіперкератозом шкіри).
Ураження слизових оболоноктрапляється нечасто, проявляється кон’юнктивітом, стоматитом, фарингітом і не має специфічних рис.
Суглобовий синдром не є провідним у клінічній картині дерматоміозиту/ поліміозиту, його спостерігають менше ніж у третини хворих. Проявляється суглобовий синдром переважно поліартралгіями, ймовірний розвиток симетричного артриту кистей, рідше — ліктьових, плечових, колінних суглобів. Артрит швидко зникає після призначення глюкокортико’їдів, не призводить до деструкції і деформації суглобів. Обмеження функції буває лише за рахунок м’язових контрактур.
У 10—30% хворих переважно при дерматоміозиті, антисинтетазному синдромі, виникає синдром Рейно. Зазвичай він не є вираженим, має двофазний характер і не супроводжується трофічними розладами.
При дерматоміозиті/поліміозиті досить часто (у 20—30% хворих) виникають ураження серця, у більшості випадків вони мають безсимптомний перебіг. Основними видами патології є міокардит і міокардіофіброз. Міокардит найчастіше проявляється помірною тахікардією і незначними відхиленнями на ЕКГ (зниження вольтажу, зміни зубця 7). Задишка та інші ознаки серцевої недостатності з’являються в разі розвитку тахіаритмії і порушень провідності.
Основні причини задишки у хворих на дерматоміозит/поліміозит пов’язані із залученням до патології дихальної системи. Часто внаслідок слабкості дихальних м’язів, насамперед міжребрових і діафрагмових, виникають порушення вентиляції, що проявляється частим поверхневим диханням та інс- піраторною задишкою. Гіповентиляція і аспірація рідини та їжі при дисфагії спричиняють розвиток пневмоній — основної причини летальності при ідіопатичному дерматоміозиті/поліміозиті. У 10% хворих спостерігають інтерстиційне ураження легень. Часом воно проявляється в дебюті захворювання як гострий дифузний альвеоліт: характерні гарячка, виснажливий непродуктивний кашель, швидко прогресує легенева недостатність, аускультативна картина бідна (жорстке дихання в нижніх відділах, можлива крепітація на обмежених ділянках). Частіше перебіг буває субклінічним, і лише при інструментальному обстеженні виявляють ознаки інтерстиційної пневмонії з переходом у базальний пневмофіброз або фіброзивний альвеоліт, що повільно прогресує. Наслідком обох патологічних процесів інколи буває легенева гіпертензія.
Оцінюючи легеневу патологію, завжди слід пам’ятати про ймовірність первинного пухлинного чи метастатичного процесу, а також активізації туберкульозної інфекції на тлі масивної терапії глюкокортикоїдами.
Органи травлення залучаються майже в половини хворих. Найбільше клінічне і діагностичне значення мають порушення ковтання, зумовлені зниженням скоротливої здатності м’язів м’якого піднебіння, язика, гортані і верхньої третини стравоходу. Нерідко проявами хвороби є анорексія, біль у животі. Інколи виявляють гепатомегалію, ерозивно-виразковий гастрит або ентероколіт, а також ускладнення виразок — кровотечі та перфорації, в основі яких лежить васкуліт по ходу травного тракту. На тлі лікування можуть виникати гастроентерологічні ускладнення фармакотерапії.
Ураження нирок не є типовим для дерматоміозиту/поліміозиту. Дуже рідко розвивається нефрит із сечовим синдромом, як казуїстичні випадки — з нефротичним синдромом. Наявність у хворого з доведеним поліміозитом клінічно значущої патології нирок потребує особливо ретельного підходу до встановлення діагнозу і виключення насамперед перехресних синдромів — комбінації дерматоміозиту/поліміозиту із СЧВ або ССД.
Специфічних уражень нервової системи не відзначено, трапляються окремі випадки поліневриту, частіше при дерматоміозиті/поліміозиті виявляють вегетативні дисфункції. У разі тяжкого перебігу хвороби можливі гіпофункція гонад, надниркових залоз.
Лабораторні дані
Суттєві зміни в загальному аналізі крові не характерні. У частини хворих виникають помірна анемія, лейкоцитоз із незначним зсувом формули вліво, рідше відзначають лейкопенію, еозинофілію. Лише у 50% хворих помірно збільшується ШОЕ, у решти вона залишається незмінною. Значне збільшення ШОЕ може спостерігатись у хворих з ураженням легень і паранеопластичним дерматоміозитом/поліміозитом. Підвищення рівня СРП, а2- і у-глобулінів, фібриногену також не є закономірним і не завжди корелює з активністю хвороби.
Найважливішими лабораторними змінами є підвищення концентрації в крові "м’язових" ферментів — креатинфосфокінази (КФК), альдолази, лактатдегідрогенази, аланінової та, особливо, аспарагінової амінотрансфераз, а також міоглобіну. Найбільш специфічний і чутливий маркер запалення та ушкодження м’язової тканини — КФК, активність якої в більшості хворих зростає в 10 разів і більше. Рівень цього ферменту є головним лабораторним орієнтиром при оцінюванні тяжкості захворювання та ефективності лікування. Слід мати на увазі, що при дерматоміозиті/поліміозиті підвищується концентрація не тільки загальної КФК, а й її МВ-фракції, що в даної категорії хворих відображає патологію скелетних м’язів, а не міокарда. (У разі потреби оцінити ураження серця слід визначати вміст тропоніну І). Рівень КФК може залишатися нормальним у деяких хворих з тяжкою атрофією м’язів і за наявності в крові інгібіторів активності ферменту, проте кожний такий випадок потребує прискіпливого ставлення і верифікації діагнозу іншими методами.
Нерідко відзначають гіперурикемію, креатинурію, міоглобінурію.
Імунологічні дослідження виявляють у 20—40% хворих РФ, АНФ у невисоких титрах, зниження активності комплементу, інколи — Le-клітини, що не має діагностичного значення. Важливим аргументом на користь дерматоміозиту/поліміозиту є наявність у крові високих титрів міозитспецифічних антитіл — анти-Jo-1, анти-Мі-2, анти-SRP. Для дерматоміозиту більш характерна наявність анти-Мі-2-антитіл, для поліміозиту — анти-Л-1. Зрозуміло, що відсутність цих антитіл не може бути перешкодою для встановлення діагнозу, оскільки загалом їх виявляють лише у 35—40% хворих.
У хворих із м’язовою слабкістю без шкірної висипки, особливо за нормального рівня КФК, з метою диференціальної діагностики з дисфункцією щитоподібної та надниркових залоз показано визначення Т3, Т4, ТТГ, кортизолу, а також рівня електролітів.
Як скринінгове дослідження для виключення раку передміхурової залози та яєчників доцільно визначати відповідно простатоспецифічний антиген (PSA) і онкомаркер Са-125.
Біопсію скелетних м ’язів, залучених до патологічного процесу, рекомендують проводити для підтвердження діагнозу всім хворим. Ураховуючи вогнищевість запалення, найбільш уражену зону можна визначити за допомогою магнітно-резонансної томографії або сцинтиграфії з 99тТ.
Електроміографія показана передусім у разі сумнівних результатів клінічного і лабораторного обстеження. Метод має високу чутливість при діагностиці запальної міопатії (понад 90 %), однак наявні зміни не специфічні. Нормальна електрична активність проксимальних груп м’язів дає підставу практично виключити діагноз дерматоміозит/поліміозит. Типовими змінами електроміограми при дерматоміозиті/поліміозиті є поліфазні потенціали дії м’язових волокон з низькою амплітудою і короткою тривалістю, патологічна активність міофібрил у спокої і в стані подразнення; швидкість проведення нервового імпульсу не відрізняється від норми.
Інші інструментальні дослідження. Усім хворим потрібно проводити рентгенографію легень або рентгенівську комп’ютерну томографію, у тому числі для виявлення раку легень. Показана ЕКГ, у разі нестабільних порушень ритму і провідності — добове моніторування ЕКГ. З метою пошуку пухлинних процесів за показаннями призначають рентгенологічне та ендоскопічне обстеження травного тракту, ультразвукове дослідження органів малого таза і черевної порожнини, мамографію тощо.
Гострий перебіг дерматоміозиту/поліміозиту характеризується гарячкою, генералізованим міозитом аж до повної нерухомості, еритемою, дисфагією, вісцеропатіями, за відсутності лікування — летальний кінець через 2—6 міс. За умови ранньої адекватної терапії ймовірний перехід його у підгострий чи хронічний перебіг. У разі підгострого перебігу симптоми наростають поступово, типовою є циклічність перебігу, а розгорнута клінічна картина виникає через 1—2 роки від дебюту. Хронічний перебіг є більш сприятливим, циклічним, він характеризується помірною м’язовою слабкістю та міалгіями, часом локальними. Ураження шкіри або відсутні, або проявляються у вигляді гіпер- пігментацїї, гіперкератозу. Переважають процеси атрофії і склерозу м’язів, вісцеральні прояви спостерігають рідко.
Ступінь активності оцінюють за величинами неспецифічних гострофа- зових показників (ШОЕ, СРП). Оскільки у значної частини хворих ці параметри не відбивають активності міозиту і дерматиту, до уваги береться також ступінь підвищення КФК, швидкість розгортання клінічної симптоматики, наявність вісцеропатій. Найчастіше ступінь активності відповідає гостроті перебігу захворювання.
Окремі форми дерматоміозиту/поліміозиту. У рамках класичного ідіопатичного дерматоміозиту/поліміозиту клінічний перебіг, відповідь на лікування і прогноз значною мірою залежать від наявності і різновиду міозитспецифіч- них антитіл (табл. 69). Найгірший перебіг захворювання асоціюється з наявністю антитіл до SRP, найсприятливіший — антитіл до Мі-2. Хворі, в яких виявляють антитіла до Jo-1, становлять найчисленнішу групу і, незважаючи на гостроту перебігу і раннє залучення легень, за умови проведення своєчасного інтенсивного лікування мають задовільний прогноз.
У незначної кількості хворих знаходять антитіла до PM/Scl (нуклеолярний білковий комплекс), що характерно для поєднання поліміозиту із ССД (один із варіантів перехресного синдрому).
Дерматоміозит/поліміозит, пов’язаний з пухлинами (вторинний, паранео- пластичний). Дерматоміозит/поліміозит є лідером серед паранеопластичних ревматичних синдромів. Частота виявлення пухлин при цьому захворюванні становить 20—25%, що в 10—12 разів вище, ніж у популяції. На тлі злоякісних пухлин частіше виникає дерматоміозит, ніж поліміозит. Переважання жінок або чоловіків залежить від локалізації пухлини, особливо небезпечним є вік понад 45 років. У третині випадків пухлину діагностують до появи м’язового синдрому, у решти хворих її знаходять під час обстеження з приводу міопатії, іноді це вдається зробити лише згодом. Найчастіше виявляють рак носоглотки, яєчників, легень, нирок, грудних і передміхурової залоз, шлунка, печінки. Рідше трапляються інші пухлини, гемобластози.
У жінок пухлини частіше локалізуються в малому тазі чи грудних залозах, а в чоловіків — у легенях. Підозра на паранеопластичннй дерматоміозит/поліміознт повинна виникати насамперед у хворих з типовим ураженням шкіри за відсутності чи незначної вираженості м’язової слабкості, з некротичним васкулітом шкіри. Низькою є ймовірність вторинного дерматоміозиту/поліміозиту за наявності міозитспецифічних антитіл, характерних вісцеропатій, зокрема інтерстиційного легеневого фіброзу. Пов’язаний з пухлинами дерматоміозит/поліміозит характеризується тяжким перебігом, торпідністю щодо лікування, зникненням ознак при ефективній терапії онкологічної хвороби.
Діагностика дерматоміозиту/поліміозиту базується переважно на клінічних даних і результатах біопсії м’яза. Головними клінічними змінами, які можуть бути підставою для встановлення діагнозу, є слабкість у проксимальних групах м’язів та шиї, порушення ковтання, періорбітальний геліотропний дерматит, симптом Готтрона, еритема типу шалі, декольте, на розгинальних поверхнях суглобів. Серед лабораторних даних найбільше значення має підвищення рівня КФК. Інші лабораторні тести та електроміографія мають допоміжне значення.
Діагностичні критерії дерматоміозиту/поліміозиту (К. Tanimoto et al., 1995) наведено нижче.
Шкірні критерії
1. Геліотропна висипка — пурпурово-червона набрякова еритема на повіках.
2. Ознака Готтрона — пурпурово-червона, з лущенням, атрофічна чи макульозна еритема на розгинальній поверхні суглобів пальців кистей.
3. Еритема шкіри на розгинальній поверхні ліктьових і колінних суглобів.
Критерії поліміозиту
1. Проксимальна м’язова слабкість (верхніх та нижніх кінцівок і тулуба).
2. Підвищення сироваткової КФК або альдолази.
3. М’язовий біль спонтанний чи під час пальпації.
4. Міогенні зміни на електроміограмі (поліфазні потенціали короткої тривалості, спонтанні потенціали фібриляції).
5. Виявлення анти-Jo-l -антитіл.
6. Недеструктивні артрити чи артралгії.
7. Ознаки системного запалення (гарячка понад 37 °С, підвищення концентрації СРП чи збільшення ШОЕ понад 20 мм/год за методом Вестергрена).
8. Дані мікроскопії біопсійного матеріалу, що відповідають запальному міозиту (запальна інфільтрація скелетних м’язів із дегенерацією чи некрозом міофібрил, ознаки активного фагоцитозу чи активної регенерації).
Для діагнозу дерматоміозиту потрібні принаймні один із 3 шкірних критеріїв і 4 з 8 критеріїв поліміозиту.
Для діагнозу поліміозиту потрібні принаймні 4 з 8 критеріїв поліміозиту.
Диференціальна діагностика
Хоча симптоматика дерматоміозиту/поліміозиту досить характерна, у разі переважання в клінічній картині того чи того синдрому можуть виникати діагностичні труднощі. Серед неревматичних захворювань найчастіше помилково встановлюють діагнози дерматологічного (набряк Квінке, еритродермія, алергійний контактний дерматит, бешиха тощо) та неврологічного (поліомієліт, myasthenia gravis, бульбарний чи псевдобульбарний синдром, поліневрит) профілю. Вирішальне значення в диференціальній діагностиці зі шкірними хворобами мають м’язовий синдром, який рано чи пізно долучається до шкірної висипки, специфічна локалізація останньої на кінцівках, артралгії, відсутність ефекту від лікування.
Дерматит у поєднанні з міалгіями, артралгіями, гарячкою та схудненням нерідко спонукають до диференціальної діагностики із СЧВ. Однак СЧВ притаманні полісиндромність, полісерозит, часте і раннє залучення нирок і центральної нервової системи, лімфаденопатія, специфічні ураження шкіри і слизових оболонок ("метелик", енантема, афтозний стоматит, хейліт); міалгії не супроводжуються слабкістю м’язів. Люпус-дерматит також часто поширюється на кисті, але він зазвичай оминає шкіру над розгинальними поверхнями суглобів пальців рук. При СЧВ повіки і носогубні складки залишаються вільними від висипки, тоді як для дерматоміозиту це одна з найхарактерніших локалізацій еритеми. Крім того, типовими для СЧВ лабораторними відхиленнями є тромбоцитопенія і лейкопенія, високі цифри ШОЕ, наявність антитіл до ДНК і Sm-антигену.
Дифузний набряк обличчя, синдром Рейно, ураження суглобів, явища дисфагії інколи вимагають виключення ССД. У таких випадках слід акцентувати увагу на виявленні проксимальної м’язової слабкості, біохімічних (КФК) та імунологічних критеріях. Для ССД не характерна еритема будь-якої локалізації. Дисфагія виникає власне після проковтування їжі, переважно твердої, їжа не потрапляє в ніс чи трахею. Синдром Рейно має тяжкий перебіг і майже завжди є першим симптомом хвороби. Якщо диференціальну діагностику проводять на пізніх стадіях, коли наявні кальцинати, то слід враховувати, що при дерматоміозиті/поліміозиті кальцинати локалізуються в м’язах та інших м’яких тканинах переважно у зоні таза, стегон, плечового поясу, а при ССД — у дистальних відділах кінцівок (пальці, передпліччя). До того ж склеродермічні кальцифікати не утворюються в м’язах.
Більш складною є диференціальна діагностика за відсутності шкірних проявів, тобто при поліміозиті. М’язовий синдром слід диференціювати з кількома групами захворювань — іншими ревматичними (ревматична полі- міалгія, системні васкуліти), нейром’язовими (м’язова дистрофія Дюшенна, myasthenia gravis, синдром Ітона—Ламберта, бічний аміотрофічний склероз), ендокринними (гіпотироз, гіпертироз, хвороба Аддісона, хвороба Кушінга, акромегалія), електролітними порушеннями (гіпокаліємія, гіпофосфатемія, гіпо- і гіперкальціємія), медикаментозними міопатіями (глюкокортикоїди, статини, D-пеніциламін, антималярійні препарати), уродженими і метаболічними міопатіями.
Міалгії при поліміозиті ніколи не домінують у картині хвороби, на відміну від інших системних ревматичних захворювань (ревматична поліміалгія, СЧВ, вузликовий поліартерит), при яких больовий синдром є провідним, а типова для дерматоміозиту/поліміозиту псевдопаралітична м’язова слабкість практично ніколи не розвивається. На ревматичну поліміалгію хворіють виключно особи похилого і старечого віку, ШОЕ збільшується до 50—60 мм/год. Хоча міалгії охоплюють ті самі проксимальні групи м’язів, проте відсутні об’єктивні ознаки їхнього ураження — пальпаторна болючість, слабкість, підвищення рівня м’язових ферментів, електроміографічні та гістологічні зміни. При вузликовому поліартериті міалгії зазвичай обмежені гомілками, одночасно чи невдовзі з’являються асиметричні неврити з руховими порушеннями, ураження нирок з артеріальною гіпертензією, некротичні зміни шкіри, ліведо.
На відміну від поліміозиту при myasthenia gravis, синдромі Ітона—Ламберта м’язова слабкість виникає епізодично, наростає під час фізичного навантаження, зменшується після відпочинку та введення прозерину; рано і часто уражаються м’язи очей.
Для групи спадкових м’язових дистрофій (Дюшенна, Беккера) слід враховувати сімейний анамнез, закономірність зниження сухожилкових рефлексів і розвитку псевдогіпертрофії м’язів. При цьому болючість, набряк м’язів, кальциноз і вісцерити відсутні.
Якщо клінічна картина представлена переважно слабкістю та атрофією м’язів, слід проводити диференціальну діагностику з неврологічними захворюваннями, пов’язаними з ураженнями мотонейронів. Ознаками, що характерні для первинного ушкодження м’язів, є симетричний розподіл слабкості у проксимальних групах м’язів, відносна збереженість сухожилкових рефлексів, відсутність порушень чутливості, неефективність прозерину. Важливе значення мають результати електроміографії: при неврологічних розладах потенціали дії м’язових волокон мають високу амплітуду і збільшену тривалість, змінюється швидкість проведення нервового імпульсу, тобто спостерігаються зміни, прямо протилежні таким при запальній міопатії.
Для виключення ендокринних захворювань і електролітних порушень мінімальний обсяг обстежень включає визначення в крові рівня ТТГ, Т3, Т4, кортизолу, калію, натрію, кальцію і фосфору. Міопатії такого походження є нестійкими, з’являються частіше під час навантаження і мають характеристики скоріше м’язової втоми, ніж слабкості.
Для глюкокортико’їдної міопатії характерні нормальний рівень КФК, відсутність змін у біоптатах м’язів, збільшення м’язової сили після зниження дози глкжокортикоїдів.
Усім хворим на дерматоміозит, особливо похилого віку, слід проводити цілеспрямоване обстеження для виявлення онкопатології.
Перебіг і ускладнення. Перебіг дерматоміозиту/поліміозиту дуже неоднорідний і залежить від варіанта дебюту, імунологічного субтипу, своєчасності діагностики і початку лікування. Найчастіше захворювання, особливо в осіб похилого віку, має тенденцію до прогресування, хвилеподібного перебігу. В осіб молодого віку можливе досягнення повної стійкої ремісії. Серед ускладнень найбільш серйозним і частим є аспіраційна пневмонія, а також банальна внутрішньогоспітальна пневмонія, передумови для якої створюють міопатична гіповентиляція та імуносупресивна терапія. Пневмонія є основною причиною летальності хворих. Імовірний розвиток порушень ритму серця і провідності, інфікування пролежнів, активізація вогнищ туберкульозної інфекції. Ускладнення з боку інших органів та систем виникають рідко.
Лікування
Основними засобами лікування є глюкокортикоїди. Залежно від гостроти перебігу і тяжкості дерматоміозиту/поліміозиту початкова доза преднізолону перорально становить від І до 2 мг на 1 кг маси тіла на добу. Протягом перших тижнів її слід уживати за 3 рази, потім — усю дозу одноразово вранці. На відміну від інших ревматичних захворювань стан таких, хворих поліпшується повільно — протягом 1—2 міс. Відсутність позитивної динаміки після 4-тижневого лікування є підставою для збільшення дози глюкокортикоїдів на 25%. Після нормалізації рівня КФК і явного наростання м’язової сили починають поступове зниження дози глкжокортикоїдів — щомісяця приблизно на 25% від попередньої дози. При цьому регулярно здійснюють клінічний контроль і не рідше 1 разу на місяць — контроль рівня
КФК. Досягнуту підтримувальну дозу — 5—10 мг на добу — можна застосовувати протягом кількох років, потім її зменшують на 1/4 таблетки щомісяця до повної відміни. Таку саму тактику застосовують і при паранеопластично- му дерматоміозиті/поліміозиті, паралельно з лікуванням онкологічної патології.
Показаннями до пульс-терапії метилпреднізолоном (по 1000 мг внутрішньовенно 3 доби поспіль) є тяжкі системні прояви (гострий дифузний альвеоліт, міокардит з ускладненнями), швидке прогресування дисфагії. В інших випадках пульс-терапія неефективна.
За наявності чинників несприятливого прогнозу до преднізолону від самого початку лікування додають цитостатичні імуносупресанти. Цитостатики також застосовують у разі поганої переносимості глюкокортикоїдів, резистентності до них або неможливості планомірного зниження дози і досягнення низьких підтримувальних доз глюкокортикоїдів. Найефективнішим є метотрексат, який призначають по 7,5—25 мг на тиждень перорально чи внутрішньовенно. Внутрішньовенний шлях уведення застосовують, якщо препарат недостатньо ефективний чи погано переноситься при пероральному вживанні. Препаратом вибору для лікування прогресивного інтерстиційного легеневого фіброзу є циклофосфан у дозі 100—200 мг на добу. При резистентних до глюкокортико’їдів формах дерматоміозиту/поліміозиту призначають циклоспорин А по 150—500 мг на добу. Азатіоприн поступається в ефективності іншим цитостатикам.
Амінохінолінові похідні (гідроксихлорохін 200 мг на добу) іноді застосовують для лікування шкірних проявів дерматоміозиту.
Ефективним методом лікування дерматоміозиту/поліміозиту, резистентного до стандартного лікування глюкокортикоїдами і цитостатиками (за винятком паранеопластичного), а також ювенільного дерматоміозиту/поліміозиту є внутрішньовенне введення імуноглобуліну. Його призначають по 2 г на 1 кг маси тіла за один день або по 1 г на 1 кг маси тіла за два дні 1 раз на місяць упродовж 3 міс.
Останнім часом при рефрактерних формах дерматоміозиту/поліміозиту в дорослих і підлітків з успіхом почали застосовувати препарати, що нейтралізують фактор некрозу пухлини (етанерцепт, інфліксимаб).
Нестероїдні протизапальні препарати показані лише для короткочасного лікування суглобового синдрому. Якщо з’являються кальцинати, проводять лікування колхіцином і комплексонами — двонатрієва сіль етилендіамінтетраоцтової кислоти (ЕДТА) — внутрішньовенно. Доказова база щодо ефективності препаратів метаболічного ряду (анаболічні стероїди, вітаміни тощо) слабка.
Метою лікувальної фізкультури при поліміозиті є запобігання розвитку контрактур і деформацій. У гострій фазі хвороби доцільно щоденно виконувати пасивні рухи в суглобах у повному обсязі, за потреби для профілактики деформацій, пов’язаних з укороченням м’язів, призначають іммобілізацію. Пізніше здійснюють перехід до активних рухів. У неактивній фазі міозиту застосовують лікувальну гімнастику, фізіотерапевтичні процедури.
Прогноз після впровадження в практику лікування глкжокортикоїдів значно поліпшився. У середньому 5-річна виживаність при ідіопатичному дерматоміозиті/поліміозиті становить 85%. Найменшою вона є у хворих з анти-SRP-антитілами (25%), найвищою — з анти-Мі-2-антитілами (90—100%). Хворі з антитілами до Jo-1 та з відсутністю міозитспецифічних антитіл займають проміжну позицію. При паранеопластичному дерматоміозиті/поліміозиті прогноз значно гірший і залежить від результату лікування пухлини. Крім імунологічних маркерів чинниками несприятливого прогнозу ідіопа- тичного дерматоміозиту/поліміозиту є похилий вік, гарячка і тяжкий міозит із дисфагією в дебюті хвороби, ураження легень, серця і травного тракту. Погіршують прогноз також пізнє встановлення діагнозу, неадекватна терапія на початку та в динаміці хвороби.
Профілактика. Головними складниками вторинної профілактики, спрямованої на недопущення генералізації процесу і подальших загострень, є якомога більш рання діагностика, своєчасне активне і довготривале лікування, уникнення дії провокаційних чинників. Важливе значення мають адекватне лікування гострої інфекції та санація вогнищ хронічної інфекції.
Подальша інформація
Завжди консультуйтеся зі своїм лікарем, щоб переконатися, що інформація, яка відображається на цій сторінці, може бути застосована до ваших особистих обставин. Інформація призначена тільки для медичних фахівців.