Клиническая картина
Семейный идиопатический нефронофтиз - заболевание, характеризующееся выраженными дегенеративными и атрофическими изменениями в почечных канальцах, особенно в их дистальных отделах, что приводит к прогрессивному фиброзу и быстрого развития почечной недостаточности. Относится к семейно-наследственных нефропатий с рецессивным типом наследования. Эту патологию еще называют ювенильным нефронофтизом, или болезнью Фанкони. Это аутосомно-рецессивный тубулоинтерстициальные кистозная дисплазия, которая не связана с полом и ген которой NPHP расположен в длинном плече 3q21 - q22 хромосомы. Морфологическим субстратом заболевания является нерегулярное истончение базальных мембран, канальцевая атрофия и дилатация, тубулоинтерстициального склероз и наличие кист преимущественно в кортикомедулярний и мозговой зонах, которые повреждают дистальные извилистые канальцы, петлю нефрона и собирательные трубочки.
Фанкони с соавторами в 1951 г. впервые описал своеобразное заболевание почек у нескольких детей, принадлежавших к двух семей, не связанных между собой родственными узами. Данные аутопсии и прижизненной пункции почек показали, что при нефронофтизи раньше и значительное поражаются канальцы, особенно их дистальные отделы. В них обнаруживают выраженные дегенеративные и атрофические изменения. Гибель и рассасывание почечной паренхимы вызывают вторичные воспалительные изменения в соединительной ткани, заканчивающиеся прогрессивным фиброзом. Клубочки сначала нормальные, а в стадии выраженного фиброза запустевают и гиалинизуються. У больных нефронофтиз обнаруживают кистозные изменения в петле нефрона, которые имеют существенное значение в развитии почечной недостаточности.
Непосредственные причины, приводящие к изменениям канальцев, не обнаружены. Фанкони высказал предположение, что нефронофтиз связан с преждевременным "износом" нефронов. При нефронофтизи первично врожденный дефект энзимных систем эпителия дистальных канальцев с последующим развитием вторичных изменений в проксимальных канальцах и клубочках.
Лица обоих полов заболевают одинаково часто. Родители больных преимущественно здоровы. Чаще всего заболевание носит семейный характер, но могут быть и спорадические случаи.
Заболевание начинается, как правило, из синдрома несахарного мочеизнурение, резистентного к питуитрина, не ранее чем через год после рождения, а нередко в возрасте 7-8 лет или позже. Полиурия сопровождается снижением относительной плотности мочи до 1,006-1,010, в случае ограничения приема жидкости она может привести к острой дегидратации, проявляется внезапным необъяснимым повышением температуры тела, диспепсическими расстройствами, судорожными сокращениями отдельных групп мышц. Полиурия сначала не сопровождается протеинурией, последняя появляется нередко через несколько лет и обычно не достигает высокой степени. Отеки отсутствуют, артериальное давление нормальное и может повышаться только в терминальной стадии. Вторым по частоте симптомом является анемия, как правило, нормохромная. Довольно часто у больных нефронофтиз обнаруживают признаки почечной остеопатии (остеопороз, расширение эпифизов, аномалия скелета, нарушение походки) и отставание в физическом развитии.
Наличие нормоклитиннои нормохромная анемия объясняют снижением еритропоетичнои функции почек. Для нефронофтизу характерны гипонатриемия, гипокальциемия, гипокалиемия, тенденция к снижению артериального давления, метаболический ацидоз. Осмолярность плазмы соответствует осмолярности мочи, плотность снижена. Уже в младшем школьном возрасте проявляют остеодистрофии и вторичный гиперпаратиреоз. Больные отстают в физическом развитии и в пропорциональной строении тела, но почти не отстают в умственном развитии.
зависимости от пораженного локуса гена выделяют три генетические и фенотипические варианты. Инфантильный (детский) нефронофтиз (NPH2) характеризуется повреждением в 9q22-q31 локусе и клинически манифестирует развитием хронической почечной недостаточности в возрасте до 3 лет. У детей находят увеличенные почки с повышенной эхогенностью и потерей кортикомедулярнои дифференциации. Клинически выявляют анемию, гиперкалиемическая метаболический ацидоз и АГ. Морфологическая картина характеризуется диффузным склерозирующей тубулоинтерстициального нефритом с микроцистною дилатацией проксимальных трубочек и капсулы почечного клубочка, наличием микроцисты в кортикальном слое почек (в отличие от медуллярного цист в старшем возрасте при NPH1 и NPH3).
В случае семейного идиопатического нефронофтизу прежде обнаруживают гипо-и изостенурия, затем устанавливают нарушения амониогенезу. Снижение клубочковой фильтрации обнаруживают позже. Уровень натрия и калия в крови обычно нормальный. Содержание калия повышается только в поздних стадиях заболевания, при формировании выраженной почечной недостаточности, когда значительно увеличивается уровень креатинина и мочевины. Довольно часто и в относительно ранние сроки болезни наблюдают гипокальциемии и гиперфосфатемии.
Течение болезни при нефронофтизи неуклонно прогрессирует и приводит к хронической почечной недостаточности, которая иногда развивается сравнительно быстро (через 2-3 года). Обычно больные доживают до юношеского возраста. Продолжительность жизни больных нефронофтиз с момента появления первых признаков болезни составляет в среднем 6 лет, максимальный срок - 22 года.
Диагностика
В начальный период диагноз установить трудно из-за схожести клинической картины нефронофтизу и несахарного мочеизнурение, поэтому заболевание обычно распознают в стадии почечной недостаточности.
В течении нефронофтизу выделяют две стадии: 1) изолированное поражение концентрационной функции почек, что проявляется синдромом почечного диабета, 2) тотальная почечная недостаточность разной степени выраженности. Такое распределение обусловливает необходимость нефрологического обследования в каждом случае питуитринрезистентного несахарного диабета.
В случае сочетания нефронофтизу с микробно-воспалительным процессом в почечной ткани заболевание часто диагностируют как хронический пиелонефрит. Особенно сложна дифференциальная диагностика нефронофтизу и пиелонефрита при минимальных проявлениях мочевого синдрома, сочетается со снижением способности почек при отсутствии нарушений показателей, характеризующих функцию почек по выведению продуктов белкового обмена (табл. 22).
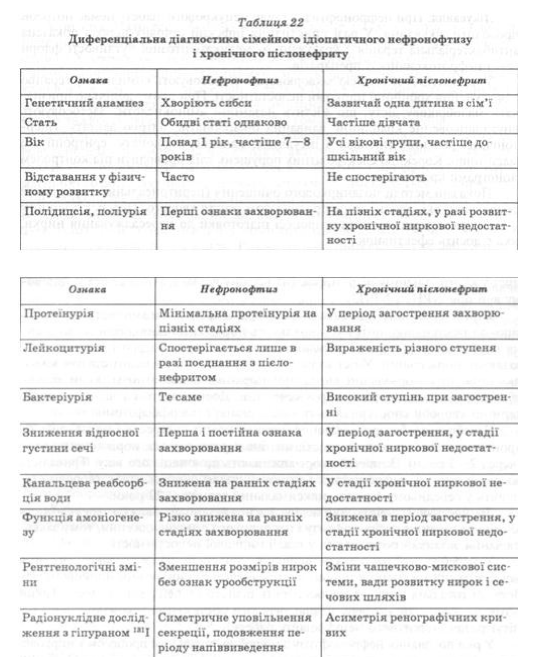
Так, устанавливая диагноз нефронофтизу, учитывают семейный анамнез, прогрессивный течение заболевания независимо от пола больных, резистентность синдрома несахарного мочеизнурение к питуитрина, наличие протеинурии и анемии.
Лечение
При нефронофтизи в стадии несахарного диабета нет необходимости проводить лечение. В случае присоединения инфекции мочевых путей показана антибактериальная терапия с учетом медикаментозной чувствительности флоры мочи и нефротоксичности препаратов.
У пациентов со II стадией заболевания применяют комплексную терапию для устранения хронической почечной недостаточности. При этом целесообразно назначать малобилкову диету, содержащую незаменимые аминокислоты. Применяют внутривенное капельное вливание бикарбонатов, натрия лактата, гипертонических растворов глюкозы с инсулином, кальция глюконата, эритроцитной массы и др.. Коррекцию электролитных нарушений следует проводить под контролем йонограмы крови и мочи.
Показаны методы внепочечного очистки (перитонеальный и хронический гемодиализ), которые, однако, лишь продлевают жизнь больного. Перспективным является применение этих методов в процессе подготовки к пересадке почки, достаточно эффективна.
Дальнейшая информация
Всегда консультируйтесь со своим врачом, чтобы убедиться, что информация, которая отображается на этой странице, может быть применена к вашим личным обстоятельствам. Информация предназначена только для медицинских специалистов.