Этиология и патогенез
Наследственный пигментный гепатоз (функциональный), или доброкачественная, гипер-билирубинемия, относится к группе заболеваний, развивающихся на фоне генетически обусловленных энзимопатий, характеризуются нарушением обмена билирубина и проявляются хронической или перемежающейся желтухой без выраженной изменения структуры и функции печени и клинических признаков повышенного гемолиза и холестаза.
зависимости от особенностей нарушения билирубинового обмена выделяют следующие формы доброкачественных гипербилирубинемий, как синдромы Жильбера, Дабина-Джонсона, Ротора и Криглера-Найяра. К пигментные гепатозы относят постгепатитный доброкачественную гипербилирубинемию, возникающий вследствие перенесенного вирусного гепатита и также генетически детерминированной.
Функциональные гипербилирубинемии часто ошибочно принимают за хронический гепатит, цирроз печени, гемолитическую желтуху, заболевания желчных путей. В последние годы с улучшением методов исследования количество случаев этих заболеваний значительно выросло, а для синдромов Жильбера и Мойленграхта (встречаются чаще всего) превысило тысячу. В настоящее время доказано, что так называемые доброкачественные (функциональные) гипербилирубинемии протекают на фоне поражения печени дистрофического характера с соответствующими ультраструктурные изменения в гепатоцитах, обусловленными генетически детерминированными энзимопатии. Основным функциональным проявлением последних является нарушение обмена билирубина в печени, а одна из форм этих заболеваний - синдром Криглера-Найяра (I тип) - характеризуется тяжелым течением и высокой смертностью. Учитывая вышесказанное оправданным является объединение этих заболеваний термином "наследственные пигментные гепатозы".
В МКБ-10 наследственные пигментные гепатозы представлены в рубрике Е80.
Е80 нарушения обмена порфирина и билирубина.
Е80.4 - синдром Жильбера.
Е80.5 - синдром Криглера - Найяра.
Е80.6 - Другие нарушения обмена билирубина.
Синдром Жильбера известен под несколькими названиями: "ювенильная перемежающаяся желтуха", "физиологическая гипербилирубинемия", "семейная негемолитич-на желтуха", "доброкачественная гипербилирубинемия" и характеризуется повышением в крови уровня непрямого билирубина.
Он был описан AN Gilbert и P.S. Lereboulet в 1900-1907 pp. под названием простой семейной желтухи, которая характеризовалась доброкачественностью, перемежающимся характером, отсутствием гемолиза и нарушением проходимости желчных путей. В 1947 p. Е. Meulengracht описал 35 случаев заболевания с такими же клиническими проявлениями. Он назвал эту болезнь ювенильной перемежающейся желтухой.
Этиология. Заболевание всегда носит наследственный характер и передается по аутосомно-доминантному типу, что обусловлено дефектом в зоне S-гена, контролирующего активность глюкуронилтрансферазы. Возникновению его способствуют такие факторы, как переохлаждение, переутомление, употребление медикаментов (например, экстракта мужского папоротника), интеркуррентные инфекции. У трети больных заболевание развивается вследствие перенесенного вирусного гепатита и также генетически обусловленным. В пользу этого утверждения есть ряд свидетельств: поражение, как и при чисто семейной форме, мужчин молодого возраста, относительная частота, значительная резистентность глюкуронидазнои активности печени, что делает маловероятным ее изолированное повреждение. Тогда говорят о доброкачественной постгепатитный гипербилирубинемию или постгепатитный форму синдрома Жильбера, понимая при этом, что вирусный гепатит играет роль фактора, который манифестирует (обнаруживает) наследственный дефект.
Патогенез. Желтуха при синдроме Жильбера преимуществу такими факторами: нарушением как захват свободного билирубина из плазмы клетками печени, так и, пожалуй, внутриклеточного транспортировки этого пигмента в гепатоцитах, что доказано исследованиями с билирубином 14 С; торможением процесса с " соединения билирубина с глюкуроновой кислотой. При этом выявляют:
• недостаточность билитранслоказы, ответственного за захват билирубина из крови и его транспортировки в гепатоцит
• дефицит Y-и Z-протеинов-лигандов (фермента глутатион-S-трансферазы), ответственных за перенос билирубина до микросом
• дефицит уридиндифосфат-глюкуронилтрансферазы (УДФГТ), обеспечивающее перенос глюкуроновой кислоты до билирубина.
Генетический дефект заключается в наличии на промоторные участке А (ТА) 6 ТАА гена, кодирующего УДФГТ, дополненного динуклеотид И, что приводит к образованию участка А (ТА) 7 ТАА.
Благодаря действию этих основных причин в крови накапливается свободная фракция билирубина, дает косвенную реакцию. Кроме этого, с помощью билирубина 51 Сг установлено, что в происхождении желтухи при синдроме Жильбера определенное значение имеет укорочение жизни эритроцитов.
время даже тяжелые поражения паренхимы печени при остром вирусном гепатите или хронических заболеваниях печени (хронический гепатит, цирроз печени) не приводят к нарушению глюкуронидазнои функции печени, что свидетельствует об очень высоком ее резерв, а снижение активности глюкуронилтрансферазы в ткани печени, сравниваемое с тем, что наблюдается при синдроме Жильбера, видзначает?? Ся при гепатоцеребральной дистрофии, а также в некоторых случаях терминальной печеночной недостаточности. Это позволяет отрицать этиологической роли вирусного гепатита в развитии синдрома Жильбера, отводя ему значение фактора, который только обнаруживает генетически обусловленную недостаточность глюкуронилтрансферазы.
Однако отождествлять этот синдром с гемолитической желтухой недопустимо, поскольку патогенез гипербилирубинемии при них принципиально разный: в случае синдрома повышенный гемолиз имеет по сравнению с другими нарушениями обмена билирубина второстепенное значение, а при гемолитической желтухе ускоренная гибель эритроцитов и повышенное образование пигмента являются ведущими патогенными звеньями. Гемолитический процесс при синдроме Жильбера носит приобретенный характер, это вторичное явление, хорошо известное при различных заболеваниях печени, протекающих с желтухой.
В некоторых случаях периодически нарушается экскреция билирубина через мембраны гепатоцитов в желчные канальцы, это приводит к повышению в крови уровня и прямого билирубина. Но этот механизм при синдроме Жильбера повреждается редко, преимущественно в случае очень длительного течения.
Патологическая анатомия
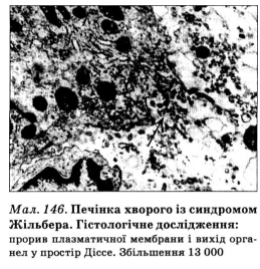
При лапароскопии микроскопических изменений печени не наблюдают. При гистологическом и гистохимическом исследовании биоптатов печени не обнаруживают никаких изменений, в других - они неспецифичны: отложение пигмента в гепатоцитах, ожирение, глюкогеноз ядер, активизация звездчатых эндотелиоцитов, в отдельных случаях - белковая дистрофия печеночных клеток и фиброз портальных полей, при этом отложения пигмента у милиарного полюса гепатоцита, преимущественно в центральных отделах дольки, нередко сочетается с активизацией звездчатых эндотелиоцитов. Структура частиц сохранена, воспалительные изменения отсутствуют. При ультраструктурном исследовании наиболее посутнишимы нарушениями являются изменения васкулярного полюса гепатоцитов (изменение формы и уменьшение числа микро-ворсинок, вплоть до полного их исчезновения, фрагментация мембраны и выход фрагментов или целых органелл в перисинусоидни пространства (пространства Диссе; рис. 146) и гиперплазия незернистые ендоцитоплазматичнои сетки печеночных клеток.
целом морфологические изменения ткани печени при синдроме Жильбера свидетельствуют об определенном дистрофическое поражение печеночных клеток, что со временем приобретает признаки Нетрудно гепатоза и является свидетельством эволюции болезни.
Клиническая картина. Первые клинические признаки синдрома Жильбера, как правило, наблюдают в молодом, подростковом или даже старшем детском возрасте и очень редко - в более поздние годы жизни или в раннем детстве. В 90% случаев синдром манифестирует в возрасте от 11 до 30 лет. Заболевания выявляют преимущественно (75-90%) у лиц мужского пола. Среди взрослых больных преобладают работники умственного труда.
Основным симптомом болезни является желтуха различной степени интенсивности. Часто наблюдают истеричность склер, гораздо реже - склер и кожи. Слизистые не иктерични. Интенсивность желтухи непостоянна. Она может значительно уменьшается или даже исчезает на длительный период, то усиливается, особенно после охлаждения, переутомления (умственной и физической), интерком-рентных заболеваний, употребления алкогольных напитков. У некоторых больных желтухи нет и заболевания выявляются после исследования крови в связи с расстройствами пищеварительной системы или благодаря профилактическим медицинским обследованием.
Примерно в 1/4 больных никаких жалоб кроме желтухи нет. В других преобладают жалобы диспепсического характера, боль в верхней части правого подреберья, особенно в период обострений. Боль в животе носит различный характер: от тупого к интенсивному приступообразного, напоминающий печеночную колику. Чаще он нерезкий, ноющая, преимущественно локализован в правом подреберье. Диспепсические нарушения проявляются тошнотой, отрыжкой, изжогой, горечью во рту и т.д.. Очень часто наблюдают астеновегетативные расстройства: подавленное настроение, слабость, снижение умственной работоспособности, потливость, головокружение, плохой сон. У некоторых больных эти жалобы появлялись лишь после того, как они узнавали о своем заболевании, что его врачи рассматривали как инфекционный или хронический гепатит. Поэтому частично субъективные симптомы можно рассматривать как проявление психогении, реактивного невроза, а не самого заболевания. С вегетативными нарушениями, очевидно, связана субфебрильная температура тела (у 15-20% больных).
Небольшое увеличение печени наблюдают примерно у 1/3 больных, она не уплотнена, безболезненна. Селезенка обычно не прощупывается.
Данные лабораторных исследований без существенных изменений. В периферической крови - нормальный или повышенное содержание гемоглобина и эритроцитов, отсутствуют изменения со стороны лейкоцитарной формулы, СОЭ не изменена или меньше нормы. Форма и диаметр эритроцитов, как правило, нормальные. Уровень билирубина повышен. У большинства больных повышается уровень свободного билирубина, а связан - либо отсутствует, либо повышенный гораздо меньше, чем свободный. Даже в период обострения болезни содержание билирубина чаще всего не превышает 85-100 мкмоль /л. У большинства больных с синдромом Жильбера наблюдают слабо выраженные изменения биохимических показателей, характеризующих функции печени, не связанные с пигментными обменом. Так, в период обострения в 1/3 больных повышается активность АЛТ в сыворотке крови, у 15% - АсАТ, ЩФ, сорбитде-гидрогеназы, печеночно-специфических изоферментов лактатдегидрогеназы (ЛДГ 4 _ 5 ) . Нередко (20-35%) наблюдается гипоальбуминемия, изменения показателей сулемовой пробы. Выраженность изменений функциональных печеночных проб зависит от длительности болезни и частоты ее обострений. В моче билирубин отсутствует, количество уробилина же бывает повышенным крайне редко и незначительно. При нук-леьидний гепатографии с бенгальским розовым 131 И выявлено небольшое нарушение поглотительной и экскреторной функции печени, с помощью пробы с бромсульфалеин относительно часто обнаруживают задержания краски (нерезко выраженное).
При дуоденальном зондировании примерно в 1/3 больных обнаруживают холангит.
Болезнь длится годами и имеет волнообразное течение. Ремиссии, бывают длинными (несколько лет), изменяются обострениями, которые проявляются усилением желтушности, увеличением размеров печени, появлением или усилением боли, диспепсических и астеновегетативных расстройств. Эти обострения чаще всего связаны с погрешностями в еде, переохлаждением, усталостью, интер-конкурентных заболеваниями. Иногда во время этих обострений связанный билирубин начинает преобладать над свободным, а в период ремиссии восстанавливаются прежние соотношения, и билирубиновой показатель, отражающий удельный вес связанной фракции, падает ниже 50%. Этот вариант определяют термином "альтернацийна форма синдрома Жильбера". Его обычно наблюдают в случае сочетания этого синдрома с инфильтративным гепатитом и воспалением желчных путей (т.е. при наличии осложнений или сопутствующих заболеваний) и характеризуется более выраженными структурными и функциональными изменениями печеночных клеток.
С годами у больных усиливается непереносимость алкоголя и жирных блюд, возрастает частота и выраженность гепато-и спленомегалии, чаще повышается активность печеночных ферментов, что свидетельствует об определенной динамике патологического процесса - нарушение функций гепатоцитов . В различные сроки (обычно через 2-5 лет от начала заболевания) могут возникнуть хронический гепатит, воспалительный процесс в желчных путях, которые определенным образом влияют на течение основного заболевания.
Диагностика . Предположение о наличии синдрома Жильбера должно быть высказано во всех случаях, когда наблюдается хроническая, нерезко выраженная, чаще интермитивна, реже постоянный желтуха, обусловленная повышением в крови преимущественно свободного билирубина, при отсутствии признаков гемолиза, выраженных нарушений общего самочувствия, функций печени и других органов и систем. Диагноз уточняют путем исследования содержания в моче глюкуроновых кислот, количество которых у больных с синдромом Жильбера снижается (менее 400 мг). Выраженное снижение глюкуронурии является патогно-моничною признаком данного заболевания.
Большое диагностическое значение имеет выявление повышенного уровня свободного билирубина в сыворотке крови у родственников пациента (в 15-40% случаев). С целью диагностики применяют также пробу с частичным голоданием. Ограничение общей суточной энергетической ценности пищи до 1,6 кДж (400 ккал) при этом заболевании ведет к подъему свободного билирубина в сыворотке крови в течение суток вдвое и выше. При гемолитических анемиях и других заболеваниях печени кратковременное частичное голодание не приводит к росту гипербилирубинемии. Первостепенное значение имеет пункционная биопсия, без которой часто невозможно отделить этот синдром от других видов гипербилирубинемии. Нормальная гистологическая структура или незначительные дистрофические изменения в ней при снижении активности глюкуронилтрансферазы гепатоцитов и выраженном увеличении непрямого билирубина в крови свидетельствует о наличии синдрома Жильбера.
Синдром Жильбера необходимо отграничивать прежде всего от гемолитических желтух, в частности от наследственных гемолитических анемий. Найпосутниши признаки последних при синдроме Жильбера отсутствуют: снижение осмотической и механической резистентности эритроцитов, изменение их морфологии и повышенная экскреция уробилиногенов тел. Надежный критерий распознавания гемолитического процесса - укорочение продолжительности жизни эритроцитов, определяемое с помощью радиоактивной метки. Условным рубежом может служить продолжительность жизни эритроцитов (т.е. период полувыведения из них 51 Сг) в течение 15 дней: меньшие показатели характерны для наследственной гемолитической желтухи и не свойственные синдрому Жильбера. Дополнительными показателями для дифференциальной диагностики служат некоторые различия этих заболеваний. Во-первых, при наследственных гемолитических желтухах железо в виде гемосидерина конечно накапливается в гепатоцитах и звездчатых эндотелиоцитах, а при синдроме Жильбера этого не происходит. Во-вторых, первые клинические проявления синдрома Жильбера в возрасте до 10 лет встречаются существенно реже, чем симптомы гемолитической желтухи. В-третьих, спленомегалию и анемию лишь изредка наблюдается при синдроме Жильбера, но они закономерны симптомами гемолитической желтухи, кроме того, у большинства больных с гемолитической желтухой степень спленомегалии и анемии выше.
Синдром Жильбера следует отличать от первичной шунтирующей гипербилиру-бинемии. Последний свойственны повышенная экскреция уробилиногенов тел, рети-кулоцитоз. Ного мозга при нормальной продолжительности жизни эритроцитов, а главное - повышение удельного веса предыдущего меченого пигмента.
Потребность дифференцировать синдром Жильбера с другими желтухи у новорожденных на практике, как правило, не возникает, поскольку в этом возрасте синдром Жильбера не манифестирует.
Хронические холестатические желтухи (внутрипеченочные и внепеченочные) в отличие от синдрома Жильбера характеризуются преимущественным повышением связанного билирубина в сыворотке крови в сочетании с клинико-лабораторных ной симптоматикой холестаза (кожный зуд, высокая активность щелочной фосфатазы в сыворотке крови, гиперхолестерин-, гипер-р-липопротеидемия и т.д..).
Дифференциальная диагностика синдрома Жильбера и таких хронических заболеваний печени, как хронический гепатит, жировой гепатоз и цирроз печени, нетяжелый. Последние протекают с более выраженными признаками поражения печени (особенно хронический алкогольный гепатит и цирроз печени) - устойчивыми гепато-и спленомегалией, изменением показателей гипоальбуминемии, гипер-аминотрансфераземии, изменением показателей коллоидных проб т.д., кроме того, им не свойственна гипербилирубинемия с преимущественным повышением свободного пигмента . В неясных случаях диагноз устанавливают с помощью пункционной биопсии печени.
При отграничении интеркуррентных острых инфекционных заболеваний у больных с синдромом Жильбера от вирусного гепатита, особенно если синдром манифестирует впервые, следует иметь в виду, что у таких больных во время любого интеркуррентного заболевания усиливается желтуха, увеличивается печень, повышается активность ферментов в сыворотке крови. В этой ситуации решающее значение имеют наблюдения в динамике (при синдроме Жильбера обнаруживают быструю нормализацию размеров печени и исчезновение признаков повреждения гепатоцитов), а также пункционная биопсия органа (отсутствие полиморфной картины острого гепатита).
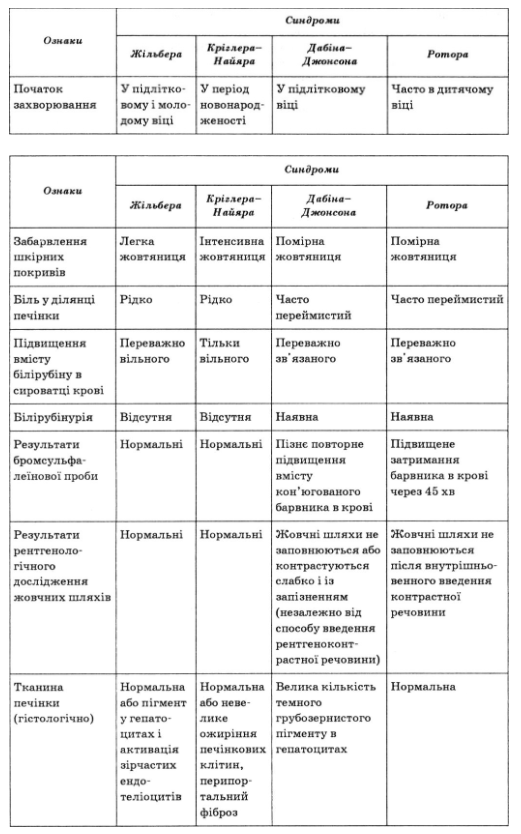
Основные дифференциально-диагностические признаки типичных форм наследственного пигментного гепатоза приведены в табл. 69.
Таблица 69
Дифференциально-диагностические признаки наследственного пигментного гепатоза
Лечение больных с синдромом Жильбера заключается в ограничении физического и психического нагрузки, рациональной диетотерапии (стол № 5) и применении препаратов - индукторов ферментов-активаторов ферментов монооксидазнои системы гепатоцитов.
частности, используют следующие препараты: а) бензонал по 0,1 г 3 раза в день в течение 2 нед б) фенобарбитал по 0,1 г 3 раза в день в течение 3 нед в) зиксорин (флумецинол) сначала по 0,4 г 3 раза в день (10 дней), а затем по 0,2 г 4 раза в день в течение 7 дней.
Общими свойствами этих соединений является их способность связываться с цитохромом Р450, липофильностью и большой период полувыведения. Под их влиянием снижается уровень билирубинемии и исчезают диспепсические явления. В процессе лечения фенобарбиталом могут возникать побочные эффекты: вялость, сонливость, атаксия. В таких случаях назначают минимальную дозу препарата (0,05 г) перед сном, что позволяет использовать его длительное время.
Для предотвращения формирования желчных (пигментных) камней рекомендуют настои из желчегонных трав, проведения ксилитного (сорбитного) или комбинированного магнезиально-ксилитного тюбажей.
Профилактика
Больные с синдромом Жильбера должны избегать значительных физических нагрузок, отказаться от курения. В их рационе, особенно во время обострения, рекомендуют ограничить жирные сорта мяса, жареные и острые блюда, пряности, консервированные продукты, запрещаются алкогольные напитки.
Во время ремиссии заболевания трудоспособность больных сохранена, снижение ее может наступить при обострении болезни.
Прогноз синдрома Жильбера благоприятный. Прогрессивные изменения в печени, которые могут привести к значительному нарушению ее функции, обычно не развиваются, однако отмечают высокую чувствительность больных к различным гепатотоксических ядов (алкоголь, некоторые медикаментозные препараты и др.).
В отдельных случаях возможны прогрессирования и эволюция заболевания в хронический гепатит, развитие воспалительного процесса в желчных путях, желчнокаменной болезни, язвы двенадцатиперстной кишки.
Синдром Мойленграхта до недавнего времени считали синонимом синдрома Жильбера и нередко даже употребляли термин "синдром Жильбера-Мойленграхта". Но позже было доказано, что это разные синдромы с похожей клинической картиной. их общими симптомами являются: снижение уровня билирубина после назначения активаторов микросомальных ферментов печени, возраст манифестации, интермитивний характер желтухи, уровень билирубина в крови не более 80-100 мкмоль /л за счет неконъюгированного фракций, клинические проявления в виде истеричности кожи и слизистых оболочек, диспепсии, астении. Но при синдроме Мойленграхта отмечают только изолированное снижение активности УДФГТ, а мембрана гепатоцита активно участвует в захвате билирубина. Лечение вроде лечения синдрома Жильбера, эффективным является назначение фенобарбитала.
Синдром Криглера-Найяра - врожденная негемолитическая неконъюгированной гипербилирубинемии, при которой уровень неконъюгированного (свободной) фракции билирубина в сыворотке крови превышает 300-340 мкмоль /л при полном отсутствии коньюго-ного билирубина через недостатисть или полную отсутствие фермента УДФГТ.
Это редкая тяжелая форма наследственной гипербилирубинемии впервые была описана S. Crigler и P. Najjar в 1952 г. у новорожденных и получила название по их именам.
Этиология и патогенез
Заболевание имеет наследственную природу с аутосомно-рецессивным характером передачи наследственного дефекта - отсутствия фермента УДФГТ (I тип), что обусловлено мутацией кодирующие гена, идентифицирован в длинной ветви хромосомы 2 (локус 2937). Резко выраженная недостаточность фермента (II тип) встречается значительно чаще и передается по аутосомно-доминантному типу.
Патогенез. Полное отсутствие в гепатоцитах глюкуронилтрансферазы, а через это и полная неспособность печени коньюгуваты билирубин (микросомальной желтуха) сопровождаются значительным повышением содержания непрямого билирубина ( синдром Криглера-Найяра I типа). Продолжительность жизни эритроцитов, т.е. продуцирование билирубина, не изменена. Выделение пигмента из организма в очень небольших количествах может происходить через стенку кишечника в их просвет, а также через альтернативные пути - образование более полярных, чем свободный билирубин, соединений, которые экскретируются с желчью и мочой. Но поскольку главный путь выведения пигмента из организма заблокирован, в крови устанавливается относительно постоянный и, как правило, достаточно высокий уровень свободного билирубина. Накапливаясь в организме, билирубин способен производить токсическое действие на ЦНС, приводить к развитию так называемой ядерной желтухи. Это закономерно случается, когда содержание его в сыворотке крови превышает 257 мкмоль /л, но может произойти при низкой концентрации свободного пигмента, если есть соответствующие факторы (ацидоз, недостаточное связывания билирубина альбумином крови вследствие гипоальбуминемии или наличии в крови веществ, которые конкурируют с пигментом за связь с белком, и др..).
При гистологическом изучении печени находят или неизмененную ткань, или небольшое ожирение гепатоцитов и незначительный портальный фиброз. Электронно-микроскопически обнаруживают изменения васкулярного полюса гепатоцитов - повреждение мембраны, уменьшение количества и сглаженность микроворсинок, выход органелл клетки в перисинусоидни пространства. Кроме того, наблюдают гиперплазию цитоплазматической сети, увеличение звездчатых эндотелиоцитов, значительное повреждение эндотелия синусоидов, расширение межклеточных пространств.
Весьма характерны изменения ЦНС - желтушная окраска подкорковых узлов с поражением нервных клеток, вплоть до их некроза (ядерная желтуха). Находят также некротические очаги в миокарде и скелетных мышцах, эрозии, кровоизлияния в слизистую оболочку желудка и кишечника. Все это следствие токсического действия свободного билирубина. Названные изменения неспецифичны для синдрома Криглера-Найяра и появляются у младенцев при различных тяжелых формах желтухи с повышением количества свободного билирубина.
Клиническими проявлениями заболевания является резко выраженная желтуха и тяжелые неврологические нарушения. Желтуха появляется, как правило, в первые дни или даже часы после рождения и сохраняется на всю жизнь. Поражения наблюдают одинаково часто у мальчиков и девочек. Кал ахолич-ный, в желчи находят лишь следы билирубина. У некоторых больных печень увеличена, спленомегалии не отмечают. Симптомы поражения ЦНС появляются в детском возрасте, иногда в первые дни жизни. Они проявляются мышечной гипертонией, нистагмом, Опистотонус, атетоз, тоническими и клоническими судорогами и т.д.. Большинство больных детей отстают в психическом и физическом развитии.
Гематологические показатели не изменены. Холецистография дает нормальные результаты.
важная биохимическая признак заболевания - повышение в сыворотке крови свободного билирубина. Билирубинемия достигает очень высокого уровня: в большинстве случаев превышает 342 мкмоль /л, а у отдельных больных доходит до 856 мкмоль /л. Билирубинурия отсутствует. Уробилиновых телец в моче и кале очень мало. Возможны периодические изменения показателей и других печеночных проб (АсАТ, содержание холестерина и коэффициент этерификации холестерина).
Большинство больных погибает на первом году жизни вследствие ядерной желтухи или интеркуррентных заболеваний. Развитие ядерной желтухи возможен и в более позднем возрасте - некоторые больные доживают до позднего детского или юношеского возраста.
Распознавание синдрома Криглера-Найяра основывается на следующих характерных признаках: выраженная постоянная или нарастающая желтуха, обусловленная повышением уровня свободного пигмента в крови, появляется в первые дни после рождения, незначительное и непостоянное нарушение других функциональных проб печени; слабое контрастирования желчных путей и пузыря контрастным веществом; темный цвет печени (накопление пигмента в гепатоцитах), присоединение к желтухе в разные сроки неврологических нарушений; семейный характер заболевания. Снижение коньюгацийнои функции печени может быть установлено при обследовании родственников пациента - гетерозиготных носителей дефектного гена. Точно диагноз обосновывают результаты биохимического исследования биоптатов печени (отсутствие активности глюк?? Ронилтрансферазы).
От описанной формы заболевания, определенной ныне как синдром Криглера-Найяра I типа, следует отличать более доброкачественное вариант, который диагностируют чаще - синдром Криглера-Найяра II типа, близкий к синдрому Жильбера . В основе его лежит генетически обусловленная недостаточность (но не отсутствие) глюкуронилтрансферазы, унаследованная по аутосомно-доминант-ным типом. Заболевание проявляется желтухой обычно на первом году жизни. Неврологические нарушения встречаются редко. Желтуха обусловлена повышением в сыворотке крови свободного пигмента, при этом уровень билирубинемия не превышает 340 мкмоль /л, а в сыворотке крови обнаруживают небольшое количество моноглюкурониду. Билирубин в моче отсутствует. В отличие от синдрома Криглера-Найяра I типа в желчи имеющийся связан пигмент. В биоптатах печени - снижение активности глюкуронилтрансферазы.
Синдром Криглера-Найяра нужно отделять от других видов желтухи новорожденных, обусловленные повышением свободного билирубина в крови.
Физиологическая желтуха новорожденных является доброкачественной транзиторной гипербилирубинемией. Она обусловлена, вероятно, тем, что коньюгацийна система печени не успевает созреть до момента рождения плода. Желтуха появляется на 1-3-й день жизни, достигает максимума к 4-5-го дня и проходит в течение 7-10 дней без лечения. Иногда, особенно у недоношенных детей, накопление в организме билирубина приводит к нарушениям ЦНС. Физиологическая желтуха новорожденных с большой вероятностью исключается при наличии любого из следующих признаков: выявление желтухи в первые 24 ч после рождения уровень гипербилирубинемии, превышающей 206 мкмоль /л у родившихся вовремя, и 257 мкмоль /л - у недоношенных; сохранение желтухи после 8-го дня жизни в родившихся вовремя, и 14-го дня - у недоношенных детей.
Мимолетная семейная гипербилирубинемия новорожденных - транзиторная, обычно доброкачественная желтуха. Появляется в первые дни жизни и сохраняется в течение 2-3 нед. Другие признаки патологического процесса чаще всего отсутствуют, хотя возможно развитие ядерной желтухи. Заболевание связывают с наличием в сыворотке крови ребенка полученной ею от матери ингибитора конъюгации билирубина. Желтуха развивается у всех детей, родившихся в данной матери.
Желтуха, связанная с грудным вскармливанием, - редкое заболевание. Оно характеризуется повышением свободного билирубина в сыворотке крови до 257-342 мкмоль /л, что наступает на 2-3-й неделе жизни и быстро (в течение нескольких дней) исчезает после прекращения грудного вскармливания. Причина желтухи неизвестна. Предполагают, что в молоке матери содержатся факторы, которые тормозят конъюгации билирубина в гепатоцитах (микросомальной желтуха). Заболевание может иметь семейный характер (поражать нескольких детей от одной матери).
Бывают желтухи новорожденных, обусловленные усиленным гемолизом различного происхождения - изоиммунизацию матери вследствие несовместимости групп крови матери и ребенка, наследственным микросфероцитоза, недостаточностью глюкозо-6-фосфатдегидрогеназы в эритроцитах и др.. У больных находят признаки повышенного гемолиза (анемия, ретикулоцитоз, увеличение выделения уробилиновых тел т.п.) вместе с признаками названных заболеваний (несовместимость групп крови матери и ребенка, изменение имуногематологичних показателей, морфологические и Энзы-мологични изменения эритроцитов и др..).
Лечение. Важным показателем лечебных мероприятий при синдроме Криглера-Найяра является снижение билирубина в крови до уровня, который не вызывает повреждения ЦНС.
эффективная фототерапия - систематическое облучение пациента лампой синего света (необходимая оборона глаз). Подобный эффект дает обычный дневной или прямой солнечный свет. Механизм светового облучения заключается в распаде билирубина до более водорастворимых и менее токсичных дериватов (ди-и монопиролы), выделяемых из организма с желчью и мочой. Лечение проводят пожизненно. Продолжительность сеансов индивидуальная (в некоторых случаях - до 15 ч в сутки). Возможные побочные эффекты - дегидратация, учащение стула, острый гемолиз. Учитывая разную биологическое действие света на нейроэндокринную систему, в фототерапии следует прибегать лишь при наличии угрожающе высокого уровня свободного билирубина в сыворотке крови. Продолжительность сеансов фототерапии может быть уменьшена путем одновременного назначения веществ, которые связывают билирубин в кишечнике и способствуют выведению из организма с испражнениями (билигнин, холестирамин).
Больным с резкой гипербилирубинемией (более 342 мкмоль /л у родившихся вовремя, и свыше 257 мкмоль /л у недоношенных) показана срочная обменная гемотрансфузия, что дает временный эффект. Лечение барбитуратами неэффективно.
Лечение ензиминдукованимы агентами, прежде барбитал, дает эффект у больных с синдромом Криглера-Найяра II типа. Механизм действия препарата связан преимущественно с увеличением активности глюкуронилтрансферазы. Лечебная доза - 3 мг на 1 кг массы тела (у детей - до 10 мг /кг) ежедневно вечером. Употребление фенобарбитала (начало действия - через 48 ч) нормализует уровень билирубина в сыворотке крови в течение 2-4 нед. Отмена препарата сопровождается постепенным повышением уровня билирубина. Имеются данные о схожий эффект оротовой кислоты (20 мг калия оротата на 1 кг массы тела в день 2-3 раз).
Показаны препараты, связывающие неконъюгированный билирубин в сыворотке крови (билигнин по 5-10 г 3 раза в день за 30-40 мин до иди, запивая водой; холестирамин по 4 г 2-4 раза в день, запивая водой).
Прогноз. Заболевания (особенно I типа) протекает тяжело и заканчивается летально в результате развития ядерной желтухи или интеркуррентных болезней.
Синдром Дабина-Джонсона - наследственная семейная идиопатическая желтуха с неидентифицированным пигментом в печеночных клетках и меланозом печени - впервые был описан TN Dabin и G.D. Johnson в 1954 г. Заболевание довольно редкое - его обнаруживают лишь в 0,3% лиц с различными заболеваниями печени.
Этиология. У 1/3 больных установлен семейный характер заболевания с аутосомно-рецессивным типом наследования. Вероятной причиной болезни является дефект гена, который контролирует синтез белкового транспортера билирубина (сМОАТ-англ., canalicular Multi Organic Anien Transporter), ответственного за выделение конъюгированного билирубина в желчный капилляр.
Главное звено патогенеза - парциальное нарушение экскреторной функции гепатоцитов (постмикросомальна гепатоцеллюлярная желтуха). Дефект этой функции распространяется на выделение из клетки билирубина, холецистографичних рентгеноконтрастных средств, бромсульфалеин, бенгальского розового, индоцианин зеленого и других органических анионов, но не распространяется, например, на желчные кислоты. Нарушение проявляется хроническим повышением в сыворотке крови связанного билирубина, а также изменением показателей некоторых функциональных проб.
Вследствие нарушения экскреторной функции гепатоцита в клетке накапливаются определенные субстраты, что, вероятно, и лежит в основе образования характерного для синдрома Дабина-Джонсона внутришньогепатоцитного пигмента.
Коньюгацийна функция печени при синдроме Дабина-Джонсона не нарушена. Продуцирования билирубина в организме не изменено.
Макроскопически печень характеризуется прежде необычным цветом с преобладанием голубого, зеленого, серого и черного тонов, интенсивность и оттенки которых бывают разными - от голубовато-зеленого до почти черного - за счет накопления меланиноподибного пигмента (меланоз).
При микроскопическом изучении ткани печени в цитоплазме гепатоцитов находят значительное количество перибилиарно расположенного пигмента. Это вместе с изменением цвета печени - характерная морфологический признак синдрома Дабина-Джонсона. Пигментные включения обычно желто-коричневого цвета, округлые, крупнозернистые, расположены преимущественно в центре долек. Количество пигмента в гепатоцитах больных с синдромом Дабина-Джонсона характеризуется большим постоянством в течение длительного времени (года). Пигмент, основой которого является меланин, исчезает при развитии острого Интерком-рентного гепатита.
Другие изменения ткани печени, которые проявляют гистологически, не характерные для синдрома Дабина-Джонсона.
Электронно-микроскопически в гепатоцитах выявляют относительно крупные (диаметром от 0,7 до 3 мкм) пигментные гранулы круглой, овальной или неправильной формы. Пигментные включения неоднородны: в них находят матрикс, состоящий из глобул высокой электронной плотности и мелких електроннощиль-ных частиц. Включение окружены одноконтурной мембраной. Другие изменения - повреждения плазматической мембраны синусоидального полюса гепатоцита (уменьшение числа микроворсинок, выход клеточных органелл в перисинусоид-ный пространство), незначительное увеличение незернистые эндоплазматической сети, уменьшение зернистой эндоплазматической сети, уменьшение числа микроворсинок желчных капилляров и нерезко выраженное расширение их просвета.
Первые клинические проявления заболевания могут появиться в период от рождения до 40-летнего возраста (в 90% случаев - до 25 лет). Синдром Дабина-Джонсона чаще диагностируют у мужчин.
Главный симптом - хроническая, или интермитивна, желтуха обычно выражена нерезко. Легкое или умеренное увеличение печени обнаруживают у большинства больных. Периодически наблюдают светлый кал и темную мочу.
У 1/3 больных синдром протекает без субъективных признаков. В остальных случаях жалобы разнообразные, чаще всего - повышенная утомляемость, боль в животе, тошнота и сниженный аппетит. Как и при синдроме Жильбера, больные предъявляют жалобы, характерные для невроза и вегетативной дистонии.
Гематологические показатели не изменены. Дуоденальное зондирование обычно дает нормальные результаты. При холецистографии и внутривенная холеграфия желчный пузырь и протоки контрактуються слабо и с опозданием (или не контрастируются вовсе).
важная биохимическая признак - гипербилирубинемия. Чаще всего она обусловлена преимущественным увеличением количества связанного пигмента (колебания в пределах 25-90%). Общий содержимое билирубина, как правило, невысок - 17-51 мкмоль /л, в отдельных случаях превышает 85 мкмоль /л. Наблюдаются билирубинурия, повышенное выделение уробилиногена с мочой.
Характерные показатели бромсульфалеиновои пробы. Обычно в крови обнаруживают незначительное задержания краски течение первых 30-45 мин, а затем характерный подъем ее, обусловлен нарушением экскреции и регургитации из гепатоцита связанного бромсульфалеин. После нагрузки билирубином такойже наблюдают позднее повторное повышение количества связанного пигмента в крови.
Изменения показателей других печеночных проб малохарактерны.
Течение синдрома хронический, благоприятный. Обострение выражено связанные с действием различных провокационных факторов - интеркуррентных инфекций, физического перенапряжения, употребления алкоголя, с менструацией, беременностью, операцией, травмой, применением оральных контрацептивов, анаболических стероидов. Эволюция процесса с развитием хронического гепатита не установлена. Время характерно сочетание синдрома Дабина-Джонсона с желчно-каменной болезнью; последняя может расцениваться, вероятно, как осложнение основного заболевания.
Распознавание синдрома Дабина-Джонсона основывается на таких характерных для него симптомах: хроническая или интермитивна желтуха, обусловленная конечно преимущественным повышением содержания конъюгированного билирубина позднее повторное повышение содержания бромсульфалеин или билирубина в крови после нагрузки этими субстратами; незначительное и непостоянное нарушение других функций печени, отсутствие контрастирования или более позднее и слабое заполнение желчных путей и пузыря контрастным веществом при рентгенологическом исследовании; темный цвет печени вследствие накопления пигмента в гепатоцитах.
Следует иметь в виду, что на практике чаще, чем типичные случаи синдрома Дабина-Джонсона, случаи с редуцированной симптоматикой, т.е. отсутствием определенных характерных признаков - измененных показателей бромсульфалеиновои пробы, отрицательных результатов холецистографии, пигмента в гепатоцитах, а также отсутствием желтухи при наличии характерного пигмента в печени. Здесь диагноз можно считать вероятным только при выявлении других характерных признаков синдрома (измененные показатели бромсульфалеиновои пробы и др.)..
Синдром Дабина-Джонсона следует отделять от других желтухи, обусловленных повышением связанного билирубина в крови, т.е. от других пост-микросомальных гепатоцеллюлярной, постгепатоцелюлярних печеночных, а также подпеченочной желтухи. Отграничение заболеваний, которым присущи эти формы желтухи, от синдрома Дабина-Джонсона на практике обычно не представляет трудностей, поскольку для первых характерны симптомы цитолиза гепатоцитов и (или) общего (не парциального!) Холестаза различной степени выраженности. В сложных случаях решающее значение имеют результаты лапароскопии, гистологического исследования биоптатов печени, иногда - эндоскопической или чрескожной черезпечинковои холл ангиографии.
Лечение синдрома Дабина-Джонсона не разработано. Больным рекомендуют избегать факторов, которые приводят к обострению процесса (употребление алкоголя и т.д.). Показаны курсы гепатопротекторных препаратов (эссенциале Н, ливолин, легалон и его аналоги, гептрал, цитраргинин, витамины группы В, липоевая кислота и др.), профилактика образования желчных камней.
Прогноз благоприятный.
Синдром Ротора. В 1948 p. А.В. Rotor описал случаи хронической семейной доброкачественной Негемолитические желтухи с повышенным уровнем конъюгированного (прямого) билирубина и нормальной гистологической структурой печени (без неидентифицированного пигмента в печеночных клетках). В дальнейшем этот феномен был назван синдромом Ротора. Описано сочетание синдромов Ротора и Дабина-Джонсона в одних и тех же семьях, что позволило некоторым авторам считать оба синдромы разновидностями одной болезни. Однако, учитывая целый ряд этиопатогенетических и клинических разногласий, большинство клиницистов рассматривают названные синдромы отдельно.
Заболевание имеет наследственную природу (почти в 1/3 описанных случаев установлен семейный характер заболевания) с авто-сомно-рецессивным путем передачи.
Главное звено патогенеза, как и при синдроме Дабина-Джонсона - нарушение экскреторной функции гепатоцита (постмикросомальна гепатоцеллюлярная желтуха), однако меньше выраженное (меньше диффузность, чем при синдроме Дабина-Джонсона) . Оно не распространяется, например, на выделение из гепатоцита рентгеноконтрастных веществ. При нагрузке бромсульфалеин проявляют повышенное задержания неконъюгированного субстрата в крови (без повторного повышения, характерного синдрома Дабина-Джонсона). Изучение динамики билирубинемии после внутршньовенного введения пигмента показывает, что вывод из крови как свободного, так и связанного пигмента существенно замедлено, что свидетельствует о соединенное нарушения функции захвата и экскреции гепатоцитами билирубина. Продуцирования билирубина, как правило, не изменено, снижение коньюгацийнои способности печени не наблюдается.
Макроскопически печень не изменена. При гистологическом исследовании ткани печени патологических изменений также не находят. Электронная микроскопия в гепатоцитах оказывает пигментные включения диаметром 0,4-1 мкм, повреждения плазматической мембраны синусоидального полюса, появление гигантских митохондрий и другие неспецифические изменения.
Первые клинические проявления синдрома Ротора нередко появляются уже в детском возрасте. Заболевание одинаково часто поражает лиц мужского и женского пола.
Основной клинический симптом - нерезко выраженная хроническая желтуха. У части больных печень несколько увеличена в размерах. Периодически наблюдают потемнение мочи.
Субъективный состояние больного нарушается реже и в меньшей степени, чем при синдроме Дабина-Джонсона. Характерные жалобы - повышенная утомляемость, боль в правом подреберье, диспепсические расстройства и снижение аппетита.
Гематологические показатели, как правило, не изменены. Дуоденальное зондирование, пероральная холецистография дают нормальные результаты. Время при введении контрастных веществ нередко не достигается рентгенологическая визуализация желчных путей.
Общее содержание билирубина в среднем выше, чем при синдроме Дабина-Джонсона. Обычно он составляет 68-103 мкмоль /л, но нередко превышает 171 мкмоль /л; 30-80% (чаще 50% и более) билирубина приходится на связанную фракцию. Наблюдают билирубинурия - повышенное выделение уробилиногена с мочой. Изменения показателей других печеночных проб (кроме отмеченных выше тестов нагрузок) не характерны.
Течение синдрома Ротора благоприятный. Заболевание длится много лет, будучи, видимо, полностью совместимым с нормальной жизнедеятельностью. Обострение возможны при воздействии тех же провокационных факторов, что и при синдроме Дабина-Джонсона (исключение - беременность, при которой нередко наблюдают даже уменьшение желтухи). Возможное осложнение - желчнокаменная болезнь.
Распознавание синдрома Ротора базируется на следующих признаках: хроническая или интермитивна желтуха, обусловленная преимущественным повышением связанного билирубина в сыворотке крови, с незначительным и непостоянным нарушением функций печени, не связанных с обменом билирубина и отсутствием видимых изменений ткани печени при световой микроскопии. Определенное значение имеют отрицательные результаты внутривенной холеграфии и повышенное задержания бромсульфалеин в крови. Синдром Ротора диагностируют в основном путем исключения других заболеваний печени (тех самых, с которыми нужно дифференцировать синдром Дабина-Джонсона). Решающее значение для диагностики и дифференциальной диагностики имеет морфологическое исследование ткани печени.
Методы лечения - см.. синдром Дабина-Джонсона.
Прогноз благоприятный, течение длительное. Развитие хронического гепатита не наблюдают.
Дальнейшая информация
Всегда консультируйтесь со своим врачом, чтобы убедиться, что информация, которая отображается на этой странице, может быть применена к вашим личным обстоятельствам. Информация предназначена только для медицинских специалистов.