Этиология и патогенез
Наследственная нефропатия - диффузное поражение почек, которое наблюдают у нескольких членов одной семьи с доминантным или рецессивным типом наследования болезни.
Первое сообщение о наследственной нефропатию принадлежит W. Dickinson (1881), который обнаружил гематурию и протеинурию у нескольких членов одной семьи и трактовал заболевания как семейный нефрит.
В группу наследственных нефропатий входят нефритоподибни синдромы, весьма схожи по клинической картиной с приобретенными заболеваниями почек - хроническим гломерулонефритом или хроническим пиелонефритом (наследственный нефрит, семейный идиопатический нефронофтиз), - и хронические тубулопатии, обусловлены генетическим дефектом транспортировки органических соединений и электролитов в почечных канальцах, вследствие чего возникают различные обменные нарушения в организме (почечный тубулярный ацидоз, фосфат-диабет и др.). Канальцы синдромы протекают как рахитоподобные заболевания и их чаще всего наблюдают в детском возрасте.
Наследственный нефрит. У взрослых из всех наследственных нефропатий чаще диагностируют наследственный хронический нефрит, который может проявляться только поражением почек или сочетаться с дефектом слуха.
В 1927 p. A. Alport указал на связь семейного нефрита с невритом слуховых нервов. По предложению D. Williamson (1961), наследственный нефрит, сочетающийся с глухотой, получил название "синдром Альпорта". В дальнейшем при наследственном нефрите были обнаружены также поражения глаз и аномалии костного скелета.
Синдром Альпорта (наследственный нефрит) - прогрессивное генетически обусловленное нарушение клубочковой фильтрации, сопровождающееся эритроцитурией и развитием хронической почечной недостаточности. Классически известный синдром Альпорта имеет также экстраренальных признаки, а именно глухоту или нарушение зрения, которое чаще проявляется у больных женского пола. В нефрологической практике диагноз синдрома Альпорта, как правило, подозревают клинически в возрасте 5-7 лет при наличии эритроцитурии с сопутствующим высокочастотным снижением слуха и верифицируют на основании результатов нефробиопсии и генетического тестирования.
Однако большинство исследователей считают целесообразным выделить наследственный нефрит как самостоятельную нозологическую форму независимо от наличия патологии слуха, а синдром Альпорта рассматривают как клинический вариант наследственного нефрита с прогностически неблагоприятным течением болезни.
По современным данным, синдром Альпорта зачастую является причиной еритроцитурий в детском возрасте. Это связано прежде всего с внедрением генной диагностики. По данным генетических исследований, частота синдрома Альпорта значительно превышает известный нефрит с тугоухостью и достигает 1:5000-10 000 населения. Таким образом, случайно обнаруженная эритроцитурия у детей чаще всего является признаком одного из вариантов синдрома Альпорта.
Заболевание наследуется по аутосомно-доминантному типу, частично связанным с полом или зависимым от пола. Измененный ген локализуется в Х-хромосоме, больные мужчины-гетерозиготы могут передавать заболевание только дочерям, а больные женщины - большинству дочерей и сыновей. Этим объясняется тяжелое течение болезни у мужчин. Однако описаны единичные случаи передачи заболевания от отца к сыну, что не укладывается в понятие наследования, сцепленного с полом. Очевидно, мутантный ген способен к кроссинговера между гомологичными участками X-и Y-xpoмосом. Возможность ассоциации автос, несущие патогенный ген с Х-хромосомой, позволяет объяснить наличие большого количества носителей заболевания в семьях с наследственным нефритом.
Механизм развития наследственного нефрита не выяснен. Есть данные, что поражение почек и глаз, неврит слуховых нервов при этом заболевании - это следствие дефекта развития одной из цепей колагенезу IV на уровне гена.
патогенетических основ морфологических изменений при заболевании является дефицит коллагена IV типа в базальных мембранах капилляров клубочка и канальцев, что приводит к их расширению (рис. 87). Дальнейшие изменения вызывают гиалинизация клубочков и развитие почечной недостаточности.
Альфа 1, 2, 5 и 6 содержатся в эпителиальной базальной мембране, альфа 1-5 - в базальной мембране гломерул. Наличие альфа 5 в обоих видах мембран позволяет проводить диагностику на основании биопсии кожи при наличии дефекта при синдроме Альпорта.
Альфа 3 (IV) - и альфа 4 (ГУ)-цепи имеющиеся в гломерулярной и частично канальцевой базальных мембранах. Альфа 5 (IV) распределяется в гломерулярной базальной мембране, капсуле почечного клубочка (капсуле Шумлянского-Боумена), частично - в дистальных канальцев базальных мембранах. Альфа 6 (ИУ)-цепь имеющийся в капсуле почечного клубочка и частично содержится в канальцевой мембране и в начале развития капиллярных петель гломерул.
Нормальные гломерулярные капилляры фильтруют плазму через базальную мембрану, богатую альфа 3, 4 и 5-цепи коллагена IV типа. Чаще всего при синдроме Альпорта отсутствует альфа5-цепь, замещается эмбрионального незрелыми альфа 1 и 2-цепями коллагена IV типа и коллагена V и VI типа. их наличие сопровождается неконтролируемой выработкой протеаз (коллагеназ и катепсин). Последние приводят к расщеплению базальной мембраны при отсутствии укрепляющей действия альфа 5-цепи.
По типу наследования определяют синдром Альпорта доминантный, сцепленный с Х-хромосомой (мутации гена COL4A5), автосомнодоминантний (COL4A3 и COL4A4 хромосомы 2, чаще у мужчин) и автосомнорецесивний (COL4A3 или COL4A4). Выделяют также XL субтип с диффузным лейомиоматозом и мегакариоцитарную тромбоцитопенией (мутации генов, кодирующих альфа 5 и 6-цепи). Всего насчитывается более 200 различных мутаций коллагена IV типа, которые устанавливают путем генеалогического и генетического анализов.
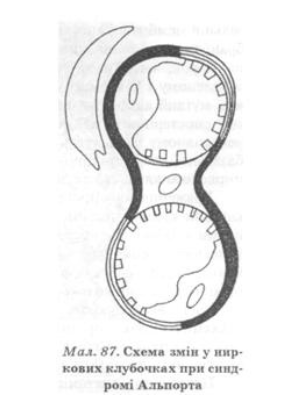
Дефекты генотипа определяются отсутствием (или уменьшением количества) коллагена. Альфа 3, 5-цепи коллагена IV типа имеющиеся, кроме гломерул, в базальной мембране капсулы хрусталика, эпителиальной мембране роговицы и мембране завитки, базальных мембранах кожи и легких. Отсюда клиническое поражение почек, глаз, уха и соединительнотканные стигмы дисембриогенезу. При сцепленных с Х-хромосомой синдроме Альпорта (80% всех случаев) обнаруживают мутации альфа 5-цепи IV типа коллагена. При диффузном лейомиоматози наблюдают почти полное отсутствие альфа С-5 (IV). При автономно-рецессивному наследовании теряются альфа 3 и 4-цепи, альфа 5 - с базальной гломерулярной мембраны, но альфа 5-6 сохраняются в капсуле почечного клубочка и дистальной канальцевой мембране.
Поражение зрения проявляются как передний лентиконус в случае повреждения капсулы хрусталика, перимакулярни дефекты, или дефекты вокруг желтого пятна сетчатки (мембрана Bruch), задняя полиморфная дистрофия (мембрана Descemet), рецидивная корнеальный эрозия (эпителиальная мембрана роговицы). Глухоту отмечают при поражении базальной мембраны улитки и непосмугованих мышечных волокон уха.
Ученые высказывают предположение, что гены мутантов, которые вызывают поражение почек и слуха, расположенные в одной хромосоме, но они могут наследоваться независимо друг от друга. Поэтому у потомков пациентов, страдающих глухотой, могут быть дети с поражением почек.
Патологическая анатомия
Патологические изменения в почках зависят от стадии, эволюции процесса и пола больного. По данным биопсии почек в ранний период болезни гистологические изменения в почечной ткани отсутствуют. В поздней стадии гистологическая картина характеризуется гломерулита - от фокально-сегментарного, минимального до диффузного, пролиферативно-мембранозного. Фибро-пластические изменения в клубочках обычно отсутствуют, склеротические явления выражены в разной степени.
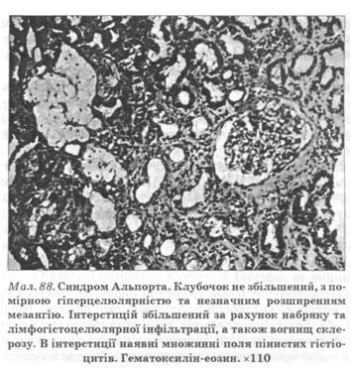
Самые тяжелые морфологические изменения обнаруживают у мужчин. Вместе с пролиферативного гломерулита выражены интерстициальная пролиферация и склероз, а также дистрофия почечных канальцев. Нередко в интерстиции обнаруживают пенистые клетки, содержащие нейтральный жир в цитоплазме и считаются патогномоничным для синдрома Альпорта (рис. 88). Однако описаны клетки наблюдают не во всех случаях, иногда их обнаруживают у больных с приобретенными заболеваниями почек, вследствие чего их специфичность для синдрома Альпорта сегодня дискутируется. Иногда при наследственном нефрите проявляют морфологическую картину, характерную для хронического пиелонефрита.
У больных, умерших от прогрессивной почечной недостаточности, на аутопсии обнаруживают уменьшение размеров почек, истончение коры, сморщивание и гиалиноз клубочков, в некоторых клубочках клеточные инфильтраты имеют вид полумесяцев, выраженная тубулярная атрофия, в интерстициальной ткани - очаги склероза и лимфоцитарные инфильтраты, склеротические изменения сосудов.
Современная клиническая классификация предусматривает наличие ювенильного (развитие хронической почечной недостаточности до 30 лет) и взрослого типов синдрома Альпорта.
Больные женского пола при наличии сцепленного с Х-хромосомой синдрома Альпорта имеют прогностически благоприятное течение заболевания. При наличии других типов наследования клиническая симптоматика существенно не зависит от пола. Таким образом, "классический" синдром Альпорта (гематурийний вариант гломерулонефрита, глухота и развитие хронической почечной недостаточности) не единственная форма заболевания, наоборот, этот вариант малораспространенным. Итак, случайно обнаруженная гломерулярная эритроцитурия нуждается прежде дифференциации с генетическими формами синдрома Альпорта. Более того, даже при наличии классического синдрома Альпорта глухота может отсутствовать.
Клиническая картина
Первые признаки поражения почек при наследственном нефрите обычно обнаруживают в детском возрасте, чаще всего в период от 3 до 10 лет. Начальные симптомы этого заболевания иногда наблюдают в возрасте до 2 лет. Клиническая картина заболевания зависит от пола больного. Во всех мужчин заболевание начинается с постоянной или обратной макро-или микрогематурии, которую может провоцировать инфекция верхних дыхательных путей. В таком случае больным ошибочно ставят диагноз острого гломерулонефрита. Обычно заболевание прогрессирует и до 30-40 лет развивается хроническая почечная недостаточность. Развитие последней до 16-летнего возраста не характерно.
У женщин заболевание протекает в легкой форме и проявляется только постоянной или обратной эритроцитурией. У некоторых больных отмечают лейкоцитурию, связанную с инфекцией мочевых путей, которая присоединяется. Развитиепочечной недостаточности не характерен. Нередко у лиц женского пола со стертым течением наследственной нефропатии мочевой синдром впервые обнаруживают в возрасте 30-40 лет во время целенаправленного обследования после выявления синдрома Альпорта у других членов семьи.
Лабораторно-инструментальные исследования. Спонтанный характер макрогематурии не исключает вероятности ее возникновения в связи с интеркуррентных заболеваниях. В промежутках между периодами макрогематурии сохраняется эритроцитурия - от 2 • 10 6 - 3 • 10 6 до 100 • 10 5 в 1 л и более, которая часто сочетается с протеинурией.
Количество белка в общем анализе мочи не превышает 1 г /л и только в период макрогематурии она увеличивается до 1,65-3,3 г /л. Суточная экскреция белка составляет 0,5-1,5 г. Протеинурия, как правило, имеет непостоянный характер.
Лейкоцитурию выявляют реже гематурия, но у некоторых больных, преимущественно женщин, она бывает значительной, чем эритроцитурия, а иногда даже сочетается с выраженной бактериурией. В таких случаях можно говорить о развитии пиелонефрита результате выборочного повышения чувствительности почечной ткани и инфекции.
О экстраренальных признаков поражения почек, то отеки и артериальная гипертензия у больных наследственный нефрит обычно отсутствуют. Только у мальчиков в возрасте 7-14 лет и у мужчин при обострении мочевого синдрома появляются пастозность тканей, артериальная гипертензия, которая в дальнейшем увеличивается по мере прогрессирования заболевания. Иногда отеки и АГ наблюдают и у женщин.
Снижение слуха значительно чаще обнаруживают у мужчин, оно проявляется преимущественно в возрасте 8-10 лет, а иногда и в более поздние сроки - в период с 11 до 15 лет и даже в 20 и 30 лет. У некоторых больных тугоухость может быть первым симптомом болезни.
На ранних стадиях заболевания снижение слуха обнаруживают лишь при аудиографии, что позволяет определить нарушение усвоения звуков с частотой 4099-2011 Гц. Тугоухость не сопровождается вестибулярными расстройствами и, как правило, прогрессирует. Вероятность проявления признаков неврита слуховых нервов в любом возрасте свидетельствует о необходимости проведения постоянного аудиометрического контроля у больных наследственный нефрит.
Поражение глаз при синдроме Альпорта случается реже, чем неврит слухового нерва, проявляется пигментным ретинитом, катарактой, сферофакиею, астигматизмом.
реже, чем глухота, но почти патогномоничным для синдрома Альпорта симптомом является передний лентиконус. На передней поверхности хрусталиков наблюдают "масляную каплю", которая может привести к нарушения рефракции и полей зрения, мешает осмотра глазного дна. У больных с синдромом Альпорта и их родственников на глазном дне вблизи плотного пятна находят также желтоватые или белые пятна.
Для наследственного нефрита характерные признаки дизембриогенезу: аномалии развития почек и мочевых путей, врожденные пороки сердца и других органов.
Диагностика
Исследование функциональной способности почек у больных с синдромом Альпорта позволяет установить, что у мужчин уже в относительно ранние сроки заболевания проявляется снижение клубочковой фильтрации по клиренсу эндогенного креатинина при нормальных величинах, характеризующих парциальные тубулярные функции почек (экскреция водородных ионов, канальцевая реабсорбция воды и др.)..
По мере прогрессирования синдрома Альпорта время со снижением фильтрационной функции почек определяют электролитные нарушения и метаболический ацидоз, степень выраженности которых зависит от стадии хронической почечной недостаточности. У больных женщин признаки почечной недостаточности могут отсутствовать в течение многих лет.
Диагностические морфологические признаки синдрома Альпорта включают пролиферацию мезангиальных клеток и матрикса, истончение стенок капилляров и тубулоинтерстициальные изменения, которые проявляют не ранее 5-7-м году жизни. В дальнейшем развиваются интерстициальный фиброз, тубулярная атрофия и дилатация, образуются пенистые клетки (вспененная цитоплазма), истончаются и удваиваются базальные мембраны клубочков и канальцев, lamina densa приобретает структуру, похожей на плетеную корзину (см. вклейку, рис. 89). С увеличением продолжительности синдрома Альпорта, который определяется генотипом больного, гломерулосклероз и интерстициальный фиброз прогрессируют, развивается хроническая почечная недостаточность. Иммунофлуоресцентного исследования на ранней стадии не информативны. Позже обнаруживают гранулярные депозиты СЗ-комплемента и иммуноглобулина М. С помощью иммунофлуоресцентного метода исследования нефробиоптату выявляют дефекты количества а-цепей коллагена, является патогномоничным признаком синдрома Альпорта.
Рентгенологическое исследование позволяет выявить некоторые особенности, характерные для наследственного нефрита. На экскреторных урограмме нередко отмечают пороки развития: удвоение лоханок, мочеточников, ротацию почки, сужение лоханочно-мочеточникового сегмента. Критерии диагноза синдрома Альпорта: наличие гематурии и нарушений слуха (зрения - за счет утончения капсулы хрусталика и переднего лентиконуса у женщин) у родственников мужского пола, развитие заболевания в 7-11-летнем возрасте, развитие нарушений функции почек, характерная картина по данным нефробиопсии ( мезангиальных пролиферация, истончение стенки капилляров, интерстициальный?? Иброз, тубулярная дистрофия и дилатация, истончение тубулярной базальной мембраны, наличие пенистых клеток) или биопсии кожи. Х-хромосомный доминантный синдром Альпорта (COL4A5), автосомнодоминантний и автосомнорецесивний синдром Альпорта (COL4A3 и COL4A4): рентгенологические изменения чашечково-лоханочной системы обычно "пиелонефритичний" характер - асимметрия заполнения мисок, их дилатация, огрубение сводов, калиектазия т.д.. У некоторых больных экскреторная урография не проявляет патологических изменений.
При цистографии в отдельных наблюдениях обнаруживают пузырно-мочеточниковый рефлюкс. Обструктивные уропатии чаще наблюдается при синдроме Альпорта.
Диагностика. Синдром Альпорта следует подозревать тогда, когда имеются такие симптомы, как эритроцитурия, почечная недостаточность и снижение (потеря) слуха. Особенно это касается тех больных, у родственников которых наблюдались случаи глухоты или заболевания почек. Кроме того, следует помнить, что эритроцитурия является главным проявлением синдрома Альпорта и возникает в начале заболевания, в детском или юношеском возрасте. Поэтому дифференциальный диагноз следует проводить со всеми заболеваниями, сопровождающимися эритроцитурией. Однако чаще синдром Альпорта приходится дифференцировать с доброкачественной семейной гематурией и гломерулонефритом с отложением IgA в клубочках (IgA-нефропатия, болезнь Берже). В табл. 20 приведена отдельные симптомы, которые можно использовать для дифференциального диагноза с этими заболеваниями, зачастую является причиной оборотной эритроцитурии.

Течение и последствия наследственного нефрита значительной мере зависят от пола больного и наличия неврита слуховых нервов. У мужчин заболевание начинается рано и неуклонно прогрессирует. В течение нескольких лет с момента выявления первых признаков заболевания наблюдают лишь мочевой синдром и тугоухость. В последующие годы появляются отеки, артериальная гипертензия, развивается глухота. Больные умирают от хронической почечной недостаточности в возрасте 15-30 лет.
При отсутствии неврита слуховых нервов и поражения глаз период компенсации у мужчин длительный. Под влиянием интеркуррентных инфекций и физической нагрузки гематурия обычно усиливается независимо от пола больного.
Течение заболевания у женщин чаще доброкачественное. Первые симптомы болезни возникают в более позднем возрасте, чем у мужчин, проявляются изолированным мочевым синдромом, нарушения слуха могут отсутствовать или длительное время определяться только с помощью Аудиометрические аппаратуры. В период беременности у больных женщин может усиливаться протеинурия, появляются отеки, артериальная гипертензия, иногда развивается почечная недостаточность. После родов экстраренальных признаки поражения почек и признаки почечной недостаточности обычно исчезают. Однако не исключена вероятность в дальнейшем прогрессирование заболевания с летальным исходом.
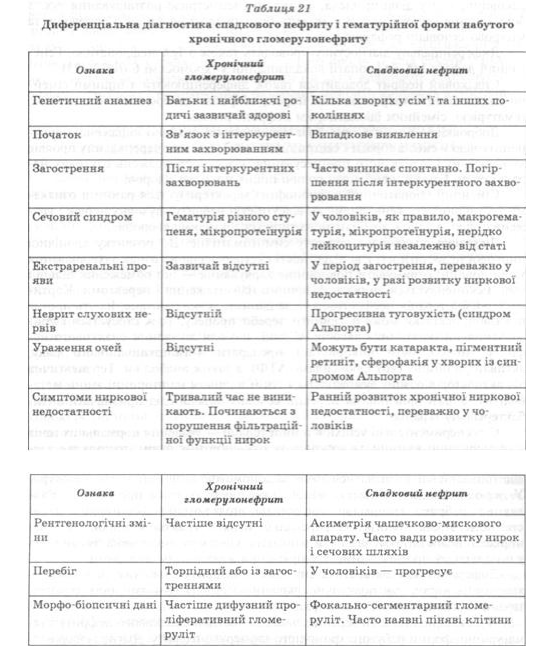
Дифференциальная диагностика наследственного нефрита и приобретенных диффузных заболеваний почек имеет практическое значение, учитывая отличие терапевтической тактики и прогноза заболевания.
сложной является дифференциальная диагностика наследственного нефрита и гематурийнои формы приобретенного хронического гломерулонефрита. Диагноз наследственного нефрита устанавливают на основании данных генетического анамнеза, спонтанного возникновения и исчезновения макрогематурии, наличии неврита слуховых нервов, прогрессивного характера заболевания у мужчин и доброкачественного его течения у женщин и других признаков (табл. 21).
При проведении дифференциальной диагностики следует помнить о синдроме брахио-оторенальний (БОР-синдром), который является автосомнодоминантним и характеризуется потерей слуха, прогрессивным развитием хронической почечной недостаточности и наличием двусторонних вдавлений в области ушной раковины, кист в области плеча, фистул или асимметрией расположения ушей. Со стороны органов мочевой системы могут быть удвоение почек и мочеточников и пузырно-мочеточниковый рефлюкс.
Дифференциальную диагностику проводят также с IgA-нефропатией. Генетический дефект IgA-нефропатии локализован в хромосоме 6 (6q22-23).
Наследственный нефрит приходится также дифференцировать с другими семейно-наследственными заболеваниями почек, в частности с доброкачественной семейной гематурией, семейным идиопатическим нефронофтизом.
Доброкачественная семейная гематурия проявляется умеренно выраженной эритроцитурией в сибсив (братья и сестры одной семьи) без экстраренальных проявлений заболевания, признаков его прогрессирования, отсутствии поражений слуховых нервов и глаз. Родители, ближайшие родственники поколениям здоровы.
Семейный идиопатический нефронофтиз характеризуется ранними признаками тубулярных поражений, анемией, отсутствием неврита слуховых нервов и поражения глаз, а также доминантного типа наследования болезни.
Лечение
Лечение наследственного нефрита симптоматическое. К развитию хронической почечной недостаточности и при отсутствии экстраренальных признаков заболевания больные должны получать полноценноепитания - без ограничения белков и соли. Рекомендуется избегать физической нагрузки и переутомления. Кортикостероидная терапия неэффективна, а по данным некоторых авторов, даже противопоказана, поскольку может ухудшить течение процесса. То же касается и назначения цитостатических средств. Есть данные, что для лечения наследственного нефрита могут быть использованы препараты 4-аминохинолинового ряда, активаторы обмена (кокарбоксилаза, АТФ), а также анаболики. Терапевтическое действие активаторов обмена обусловлено их влиянием на вторичные изменения метаболизма. В случае присоединения инфекции мочевых путей следует назначать антибактериальную терапию.
Имеются экспериментальные успехи в генной терапии - введение нормальных генов в гломерулярные клетки с помощью транспортных форм, например аденовирусных капсида (векторная терапия). Перспективным считают введение нормальных генов антенатально (специфическая генная терапия).
Трансплантация почки является очень эффективной. Антигломерулярний нефрит в пересаженной почке формируется менее 5% пациентов.
Больным наследственный нефрит показана санация очагов хронической инфекции. При наличии хронического декомпенсированного тонзиллита радикальное лечение целесообразно, несмотря на гематурию. Тонзиллэктомия противопоказана больным с признаками хронической почечной недостаточности.
При хронической почечной недостаточности по мере ее прогрессирования назначают такие же меры, как при почечной недостаточности другой этиологии.
Профилактика наследственного нефрита состоит в разъяснении больным степени достоверности риска заболевания потомства. Медико-генетической консультации особенно нуждаются больные женщины, поскольку матери передают заболевания детям.
Прогноз заболевания у женщин благоприятный. У мужчин чаще развивается хроническая почечная недостаточность. Она может возникать в разном возрасте, в разных родственников. Различают ювенильной формой заболевания, при которой у детей развивается почечная недостаточность в возрасте до 18 лет, и "тип взрослых", при котором почечная недостаточность появляется у мужчин в возрасте около 40 лет. Быстрого течения болезни и неблагоприятного прогноза следует ожидать у больных, которые унаследовали заболевания от матери и в которых обнаруживают протеинурию и значительное загрубения и расслоение базальных мембран капилляров клубочков.
Дальнейшая информация
Всегда консультируйтесь со своим врачом, чтобы убедиться, что информация, которая отображается на этой странице, может быть применена к вашим личным обстоятельствам. Информация предназначена только для медицинских специалистов.